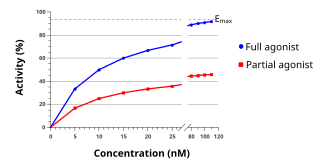
An agonist is a chemical that activates a receptor to produce a biological response. Receptors are cellular proteins whose activation causes the cell to modify what it is currently doing. In contrast, an antagonist blocks the action of the agonist, while an inverse agonist causes an action opposite to that of the agonist.

A receptor antagonist is a type of receptor ligand or drug that blocks or dampens a biological response by binding to and blocking a receptor rather than activating it like an agonist. Antagonist drugs interfere in the natural operation of receptor proteins. They are sometimes called blockers; examples include alpha blockers, beta blockers, and calcium channel blockers. In pharmacology, antagonists have affinity but no efficacy for their cognate receptors, and binding will disrupt the interaction and inhibit the function of an agonist or inverse agonist at receptors. Antagonists mediate their effects by binding to the active site or to the allosteric site on a receptor, or they may interact at unique binding sites not normally involved in the biological regulation of the receptor's activity. Antagonist activity may be reversible or irreversible depending on the longevity of the antagonist–receptor complex, which, in turn, depends on the nature of antagonist–receptor binding. The majority of drug antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on receptors.

Amoxapine, sold under the brand name Asendin among others, is a tricyclic antidepressant (TCA). It is the N-demethylated metabolite of loxapine. Amoxapine first received marketing approval in the United States in 1980, approximately 10 to 20 years after most of the other TCAs were introduced in the United States.
Cytochrome P450 3A4 is an important enzyme in the body, mainly found in the liver and in the intestine. It oxidizes small foreign organic molecules (xenobiotics), such as toxins or drugs, so that they can be removed from the body. It is highly homologous to CYP3A5, another important CYP3A enzyme.

Half maximal inhibitory concentration (IC50) is a measure of the potency of a substance in inhibiting a specific biological or biochemical function. IC50 is a quantitative measure that indicates how much of a particular inhibitory substance (e.g. drug) is needed to inhibit, in vitro, a given biological process or biological component by 50%. The biological component could be an enzyme, cell, cell receptor or microorganism. IC50 values are typically expressed as molar concentration.
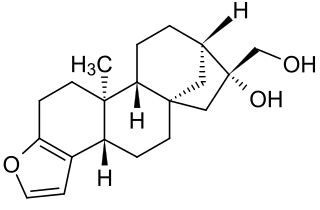
Cafestol is a diterpenoid molecule present in coffee beans. It is one of the compounds that may be responsible for proposed biological and pharmacological effects of coffee.

The bile acid receptor (BAR), also known as farnesoid X receptor (FXR) or NR1H4, is a nuclear receptor that is encoded by the NR1H4 gene in humans.

In the field of molecular biology, the pregnane X receptor (PXR), also known as the steroid and xenobiotic sensing nuclear receptor (SXR) or nuclear receptor subfamily 1, group I, member 2 (NR1I2) is a protein that in humans is encoded by the NR1I2 gene.
The constitutive androstane receptor (CAR) also known as nuclear receptor subfamily 1, group I, member 3 is a protein that in humans is encoded by the NR1I3 gene. CAR is a member of the nuclear receptor superfamily and along with pregnane X receptor (PXR) functions as a sensor of endobiotic and xenobiotic substances. In response, expression of proteins responsible for the metabolism and excretion of these substances is upregulated. Hence, CAR and PXR play a major role in the detoxification of foreign substances such as drugs.

The nociceptin opioid peptide receptor (NOP), also known as the nociceptin/orphanin FQ (N/OFQ) receptor or kappa-type 3 opioid receptor, is a protein that in humans is encoded by the OPRL1 gene. The nociceptin receptor is a member of the opioid subfamily of G protein-coupled receptors whose natural ligand is the 17 amino acid neuropeptide known as nociceptin (N/OFQ). This receptor is involved in the regulation of numerous brain activities, particularly instinctive and emotional behaviors. Antagonists targeting NOP are under investigation for their role as treatments for depression and Parkinson's disease, whereas NOP agonists have been shown to act as powerful, non-addictive painkillers in non-human primates.

The δ-opioid receptor, also known as delta opioid receptor or simply delta receptor, abbreviated DOR or DOP, is an inhibitory 7-transmembrane G-protein coupled receptor coupled to the G protein Gi/G0 and has enkephalins as its endogenous ligands. The regions of the brain where the δ-opioid receptor is largely expressed vary from species model to species model. In humans, the δ-opioid receptor is most heavily expressed in the basal ganglia and neocortical regions of the brain.

Nalorphine, also known as N-allylnormorphine, is a mixed opioid agonist–antagonist with opioid antagonist and analgesic properties. It was introduced in 1954 and was used as an antidote to reverse opioid overdose and in a challenge test to determine opioid dependence.
Herkinorin is an opioid analgesic that is an analogue of the natural product salvinorin A. It was discovered in 2005 during structure-activity relationship studies into neoclerodane diterpenes, the family of chemical compounds of which salvinorin A is a member.

RWJ-394674 is a drug that is used in scientific research. It is a potent, orally active analgesic drug that produces little respiratory depression. RWJ-394674 itself is a potent and selective agonist for δ-opioid receptors, with a Ki of 0.24 nM at δ and 72 nM at μ. However once inside the body, RWJ-394674 is dealkylated to its monodesethyl metabolite RWJ-413216, which is a potent agonist at the μ-opioid receptor and has less affinity for δ. The effect of RWJ-394674 when administered in vivo thus produces potent agonist effects at both μ and δ receptors through the combined actions of the parent drug and its active metabolite, with the δ-agonist effects counteracting the respiratory depression from the μ-opioid effects, and the only prominent side-effect being sedation.

Alazocine, also known more commonly as N-allylnormetazocine (NANM), is a synthetic opioid analgesic of the benzomorphan family related to metazocine, which was never marketed. In addition to its opioid activity, the drug is a sigma receptor agonist, and has been used widely in scientific research in studies of this receptor. Alazocine is described as a potent analgesic, psychotomimetic or hallucinogen, and opioid antagonist. Moreover, one of its enantiomers was the first compound that was found to selectively label the σ1 receptor, and led to the discovery and characterization of the receptor.

Cebranopadol is an opioid analgesic of the benzenoid class which is currently under development internationally by Grünenthal, a German pharmaceutical company, and its partner Depomed, a pharmaceutical company in the United States, for the treatment of a variety of different acute and chronic pain states. As of November 2014, it is in phase III clinical trials.

Naltalimide (INN) (code name TRK-130, formerly TAK 363) is a novel, centrally-acting opioid drug which is under development by Takeda and Toray for the treatment of overactive bladder/urinary incontinence. It acts as a potent and selective partial agonist of the μ-opioid receptor (Ki = 0.268 nM, EC50 = 2.39 nM, Emax = 66.1%) over the δ-opioid (Ki = 121 nM, EC50 = 26.1 nM, Emax = 71.0%) and κ-opioid receptors (Ki = 8.97 nM, EC50 = 9.51 nM, Emax = 62.6%). Notably, naltalimide somehow appears to lack certain undesirable side effects such as constipation seen with other μ-opioid receptor agonists such as morphine. It enhances bladder storage via suppression of the afferent limb of the micturition reflex pathway.

Thienorphine is a very potent, extremely long-acting, orally-active opioid analgesic with mixed agonist–antagonist properties which was developed by the Beijing Institute of Pharmacology and Toxicology as a potential treatment for opioid dependence. It is a high-affinity, balanced ligand of the μ-, δ-, and κ-opioid receptors, behaving as a partial agonist of the μ- and κ-opioid receptors and as an antagonist of the δ-opioid receptor. It also possesses relatively low affinity for the nociceptin receptor, where it acts as an antagonist.

Acetothiolutamide is a selective androgen receptor modulator (SARM) derived from the nonsteroidal antiandrogen bicalutamide that was described in 2002 and was one of the first SARMs to be discovered and developed. It is a high-affinity, selective ligand of the androgen receptor (AR), where it acts as a full agonist in vitro, and has in vitro potency comparable to that of testosterone. However, in vivo, acetothiolutamide displayed overall negligible androgenic effects, though significant anabolic effects were observed at high doses. In addition, notable antiandrogen effects were observed in castrated male rats treated with testosterone propionate. The discrepancy between the in vitro and in vivo actions of acetothiolutamide was determined to be related to rapid plasma clearance and extensive hepatic metabolism into a variety of metabolites with differing pharmacological activity, including AR partial agonism and antagonism. In accordance with its poor metabolic stability, acetothiolutamide is not orally bioavailable, and shows activity only via injected routes such as subcutaneous and intravenous.
Peripherally acting μ-opioid receptor antagonists (PAMORAs) are a class of chemical compounds that are used to reverse adverse effects caused by opioids interacting with receptors outside the central nervous system (CNS), mainly those located in the gastrointestinal tract. PAMORAs are designed to specifically inhibit certain opioid receptors in the gastrointestinal tract and with limited ability to cross the blood–brain barrier. Therefore, PAMORAs do not affect the analgesic effects of opioids within the central nervous system.

