Related Research Articles

The human microbiome is the aggregate of all microbiota that reside on or within human tissues and biofluids along with the corresponding anatomical sites in which they reside, including the skin, mammary glands, seminal fluid, uterus, ovarian follicles, lung, saliva, oral mucosa, conjunctiva, biliary tract, and gastrointestinal tract. Types of human microbiota include bacteria, archaea, fungi, protists, and viruses. Though micro-animals can also live on the human body, they are typically excluded from this definition. In the context of genomics, the term human microbiome is sometimes used to refer to the collective genomes of resident microorganisms; however, the term human metagenome has the same meaning.
The branches of science known informally as omics are various disciplines in biology whose names end in the suffix -omics, such as genomics, proteomics, metabolomics, metagenomics, phenomics and transcriptomics. Omics aims at the collective characterization and quantification of pools of biological molecules that translate into the structure, function, and dynamics of an organism or organisms.
The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.

Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.

Gut microbiota, gut microbiome, or gut flora are the microorganisms, including bacteria, archaea, fungi, and viruses, that live in the digestive tracts of animals. The gastrointestinal metagenome is the aggregate of all the genomes of the gut microbiota. The gut is the main location of the human microbiome. The gut microbiota has broad impacts, including effects on colonization, resistance to pathogens, maintaining the intestinal epithelium, metabolizing dietary and pharmaceutical compounds, controlling immune function, and even behavior through the gut–brain axis.
Dysbiosis is characterized by a disruption to the microbiome resulting in an imbalance in the microbiota, changes in their functional composition and metabolic activities, or a shift in their local distribution. For example, a part of the human microbiota such as the skin flora, gut flora, or vaginal flora, can become deranged, with normally dominating species underrepresented and normally outcompeted or contained species increasing to fill the void. Similar to the human gut microbiome, diverse microbes colonize the plant rhizosphere, and dysbiosis in the rhizosphere, can negatively impact plant health. Dysbiosis is most commonly reported as a condition in the gastrointestinal tract or plant rhizosphere.
The Human Microbiome Project (HMP) was a United States National Institutes of Health (NIH) research initiative to improve understanding of the microbiota involved in human health and disease. Launched in 2007, the first phase (HMP1) focused on identifying and characterizing human microbiota. The second phase, known as the Integrative Human Microbiome Project (iHMP) launched in 2014 with the aim of generating resources to characterize the microbiome and elucidating the roles of microbes in health and disease states. The program received $170 million in funding by the NIH Common Fund from 2007 to 2016.

Microbiota are the range of microorganisms that may be commensal, mutualistic, or pathogenic found in and on all multicellular organisms, including plants. Microbiota include bacteria, archaea, protists, fungi, and viruses, and have been found to be crucial for immunologic, hormonal, and metabolic homeostasis of their host.

RNA-Seq is a technique that uses next-generation sequencing to reveal the presence and quantity of RNA molecules in a biological sample, providing a snapshot of gene expression in the sample, also known as transcriptome.
Pathogenomics is a field which uses high-throughput screening technology and bioinformatics to study encoded microbe resistance, as well as virulence factors (VFs), which enable a microorganism to infect a host and possibly cause disease. This includes studying genomes of pathogens which cannot be cultured outside of a host. In the past, researchers and medical professionals found it difficult to study and understand pathogenic traits of infectious organisms. With newer technology, pathogen genomes can be identified and sequenced in a much shorter time and at a lower cost, thus improving the ability to diagnose, treat, and even predict and prevent pathogenic infections and disease. It has also allowed researchers to better understand genome evolution events - gene loss, gain, duplication, rearrangement - and how those events impact pathogen resistance and ability to cause disease. This influx of information has created a need for bioinformatics tools and databases to analyze and make the vast amounts of data accessible to researchers, and it has raised ethical questions about the wisdom of reconstructing previously extinct and deadly pathogens in order to better understand virulence.
Metaproteomics is an umbrella term for experimental approaches to study all proteins in microbial communities and microbiomes from environmental sources. Metaproteomics is used to classify experiments that deal with all proteins identified and quantified from complex microbial communities. Metaproteomics approaches are comparable to gene-centric environmental genomics, or metagenomics.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.

A microbiome is the community of microorganisms that can usually be found living together in any given habitat. It was defined more precisely in 1988 by Whipps et al. as "a characteristic microbial community occupying a reasonably well-defined habitat which has distinct physio-chemical properties. The term thus not only refers to the microorganisms involved but also encompasses their theatre of activity". In 2020, an international panel of experts published the outcome of their discussions on the definition of the microbiome. They proposed a definition of the microbiome based on a revival of the "compact, clear, and comprehensive description of the term" as originally provided by Whipps et al., but supplemented with two explanatory paragraphs. The first explanatory paragraph pronounces the dynamic character of the microbiome, and the second explanatory paragraph clearly separates the term microbiota from the term microbiome.
The microbiota are the sum of all symbiotic microorganisms living on or in an organism. The fruit fly Drosophila melanogaster is a model organism and known as one of the most investigated organisms worldwide. The microbiota in flies is less complex than that found in humans. It still has an influence on the fitness of the fly, and it affects different life-history characteristics such as lifespan, resistance against pathogens (immunity) and metabolic processes (digestion). Considering the comprehensive toolkit available for research in Drosophila, analysis of its microbiome could enhance our understanding of similar processes in other types of host-microbiota interactions, including those involving humans. Microbiota plays key roles in the intestinal immune and metabolic responses via their fermentation product, acetate.
Hologenomics is the omics study of hologenomes. A hologenome is the whole set of genomes of a holobiont, an organism together with all co-habitating microbes, other life forms, and viruses. While the term hologenome originated from the hologenome theory of evolution, which postulates that natural selection occurs on the holobiont level, hologenomics uses an integrative framework to investigate interactions between the host and its associated species. Examples include gut microbe or viral genomes linked to human or animal genomes for host-microbe interaction research. Hologenomics approaches have also been used to explain genetic diversity in the microbial communities of marine sponges.
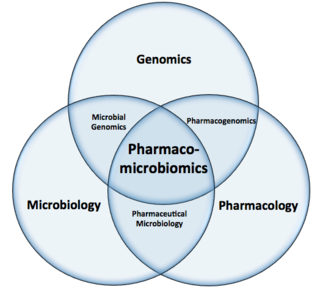
Pharmacomicrobiomics, proposed by Prof. Marco Candela for the ERC-2009-StG project call, and publicly coined for the first time in 2010 by Rizkallah et al., is defined as the effect of microbiome variations on drug disposition, action, and toxicity. Pharmacomicrobiomics is concerned with the interaction between xenobiotics, or foreign compounds, and the gut microbiome. It is estimated that over 100 trillion prokaryotes representing more than 1000 species reside in the gut. Within the gut, microbes help modulate developmental, immunological and nutrition host functions. The aggregate genome of microbes extends the metabolic capabilities of humans, allowing them to capture nutrients from diverse sources. Namely, through the secretion of enzymes that assist in the metabolism of chemicals foreign to the body, modification of liver and intestinal enzymes, and modulation of the expression of human metabolic genes, microbes can significantly impact the ingestion of xenobiotics.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.

A microbiome-wide association study (MWAS), otherwise known as a metagenome-wide association study (MGWAS), is a statistical methodology used to examine the full metagenome of a defined microbiome in various organisms to determine if some feature of the microbiome is associated with a host trait. MWAS has been adopted by the field of metagenomics from the widely used genome-wide association study (GWAS).
References
- 1 2 3 4 Filiatrault MJ (October 2011). "Progress in prokaryotic transcriptomics". Current Opinion in Microbiology. 14 (5): 579–86. doi:10.1016/j.mib.2011.07.023. PMID 21839669.
- ↑ Bashiardes S, Zilberman-Schapira G, Elinav E (2016). "Use of Metatranscriptomics in Microbiome Research". Bioinformatics and Biology Insights. 10: 19–25. doi:10.4137/BBI.S34610. PMC 4839964 . PMID 27127406.
- ↑ Whipps JM, Lewis K, Cooke RC (1988). Mycoparasitism and Plant Disease Control. Manchester, UK: Manchester University Press. pp. 161–87.
- ↑ Moran MA (2009). "Metatranscriptomics: eavesdropping on complex microbial communities". Microbe Magazine. 4 (7): 329–34. doi: 10.1128/microbe.4.329.1 .
- ↑ Apirion D, Miczak A (February 1993). "RNA processing in prokaryotic cells". BioEssays. 15 (2): 113–20. doi:10.1002/bies.950150207. PMID 7682412. S2CID 42365781.
- ↑ Peimbert M, Alcaraz LD (2016). "A Hitchhiker's Guide to Metatranscriptomics". Field Guidelines for Genetic Experimental Designs in High-Throughput Sequencing. Springer. pp. 313–342.
- ↑ Dumont MG, Pommerenke B, Casper P (October 2013). "Using stable isotope probing to obtain a targeted metatranscriptome of aerobic methanotrophs in lake sediment". Environmental Microbiology Reports. 5 (5): 757–64. doi:10.1111/1758-2229.12078. PMID 24115627.
- ↑ Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. (May 2011). "Full-length transcriptome assembly from RNA-Seq data without a reference genome". Nature Biotechnology. 29 (7): 644–52. doi:10.1038/nbt.1883. PMC 3571712 . PMID 21572440.
- ↑ Li B, Dewey CN (2011). "Rsem: accurate transcript quantification from RNA-seq data with or without a reference genome". BMC Bioinformatics. 12 (1): 323. doi: 10.1186/1471-2105-12-323 . PMC 3163565 . PMID 21816040.
- ↑ Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (August 2013). "De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis". Nature Protocols. 8 (8): 1494–512. doi:10.1038/nprot.2013.084. PMC 3875132 . PMID 23845962.
- ↑ De Bona F, Ossowski S, Schneeberger K, Rätsch G (August 2008). "Optimal spliced alignments of short sequence reads". Bioinformatics. 24 (16): i174–80. doi: 10.1093/bioinformatics/btn300 . PMID 18689821.
- 1 2 Leimena MM, Ramiro-Garcia J, Davids M, van den Bogert B, Smidt H, Smid EJ, Boekhorst J, Zoetendal EG, Schaap PJ, Kleerebezem M (2013). "A comprehensive metatranscriptome analysis pipeline and its validation using human small intestine microbiota datasets". BMC Genomics. 14 (1): 530. doi: 10.1186/1471-2164-14-530 . PMC 3750648 . PMID 23915218.
- ↑ Yost S, Duran-Pinedo AE, Teles R, Krishnan K, Frias-Lopez J (December 2015). "Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis". Genome Medicine. 7 (1): 27. doi: 10.1186/s13073-015-0153-3 . PMC 4410737 . PMID 25918553.
- ↑ Yost S, Duran-Pinedo AE, Teles R, Krishnan K, Frias-Lopez J (2015). "Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis". Genome Medicine. 7 (1): 27. doi: 10.1186/s13073-015-0153-3 . PMC 4410737 . PMID 25918553.
- ↑ Duran-Pinedo AE, Chen T, Teles R, Starr JR, Wang X, Krishnan K, Frias-Lopez J (August 2014). "Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis". The ISME Journal. 8 (8): 1659–72. doi:10.1038/ismej.2014.23. PMC 4817619 . PMID 24599074.
- ↑ Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, Whiteley M (April 2014). "Metatranscriptomics of the human oral microbiome during health and disease". mBio. 5 (2): e01012–14. doi:10.1128/mBio.01012-14. PMC 3977359 . PMID 24692635.
- ↑ Xiong X, Frank DN, Robertson CE, Hung SS, Markle J, Canty AJ, McCoy KD, Macpherson AJ, Poussier P, Danska JS, Parkinson J (2012). "Generation and analysis of a mouse intestinal metatranscriptome through Illumina based RNA-sequencing". PLOS ONE. 7 (4): e36009. Bibcode:2012PLoSO...736009X. doi: 10.1371/journal.pone.0036009 . PMC 3338770 . PMID 22558305.
- ↑ Niu SY, Yang J, McDermaid A, Zhao J, Kang Y, Ma Q (November 2018). "Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes". Briefings in Bioinformatics. 19 (6): 1415–1429. doi: 10.1093/bib/bbx051 . PMID 28481971.
- ↑ Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, Huttenhower C (November 2018). "Species-level functional profiling of metagenomes and metatranscriptomes". Nature Methods. 15 (11): 962–968. doi:10.1038/s41592-018-0176-y. PMC 6235447 . PMID 30377376.
- ↑ Martinez X, Pozuelo M, Pascal V, Campos D, Gut I, Gut M, Azpiroz F, Guarner F, Manichanh C (May 2016). "MetaTrans: an open-source pipeline for metatranscriptomics". Scientific Reports. 6: 26447. Bibcode:2016NatSR...626447M. doi:10.1038/srep26447. PMC 4876386 . PMID 27211518.
- ↑ Westreich ST, Korf I, Mills DA, Lemay DG (September 2016). "SAMSA: a comprehensive metatranscriptome analysis pipeline". BMC Bioinformatics. 17 (1): 399. doi: 10.1186/s12859-016-1270-8 . PMC 5041328 . PMID 27687690.
- ↑ Milanese, et al. (2019). "Microbial abundance, activity and population genomic profiling with mOTUs2". Nature Communications. 10 (1): 1014. Bibcode:2019NatCo..10.1014M. doi:10.1038/s41467-019-08844-4. PMC 6399450 . PMID 30833550.
- ↑ Karasov WH, Martínez del Rio C, Caviedes-Vidal E (2011). "Ecological physiology of diet and digestive systems". Annual Review of Physiology. 73: 69–93. doi:10.1146/annurev-physiol-012110-142152. hdl: 11336/14704 . PMID 21314432.
- ↑ LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M (April 2013). "Bacteria as vitamin suppliers to their host: a gut microbiota perspective". Current Opinion in Biotechnology. 24 (2): 160–8. doi:10.1016/j.copbio.2012.08.005. hdl: 11336/2561 . PMID 22940212.
- ↑ Claus SP, Guillou H, Ellero-Simatos S (2016). "The gut microbiota: a major player in the toxicity of environmental pollutants?". npj Biofilms and Microbiomes. 2: 16003. doi:10.1038/npjbiofilms.2016.3. PMC 5515271 . PMID 28721242.
- ↑ Kamada N, Seo SU, Chen GY, Núñez G (May 2013). "Role of the gut microbiota in immunity and inflammatory disease". Nature Reviews. Immunology. 13 (5): 321–35. doi:10.1038/nri3430. PMID 23618829. S2CID 205491968.
- ↑ Abreu MT (February 2010). "Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function". Nature Reviews. Immunology. 10 (2): 131–44. doi:10.1038/nri2707. PMID 20098461. S2CID 21789611.
- ↑ Sommer F, Bäckhed F (April 2013). "The gut microbiota--masters of host development and physiology". Nature Reviews. Microbiology. 11 (4): 227–38. doi:10.1038/nrmicro2974. PMID 23435359. S2CID 22798964.
- ↑ Hooper LV, Littman DR, Macpherson AJ (June 2012). "Interactions between the microbiota and the immune system". Science. 336 (6086): 1268–73. Bibcode:2012Sci...336.1268H. doi:10.1126/science.1223490. PMC 4420145 . PMID 22674334.
- ↑ Gosalbes MJ, Durbán A, Pignatelli M, Abellan JJ, Jiménez-Hernández N, Pérez-Cobas AE, Latorre A, Moya A (March 2011). "Metatranscriptomic approach to analyze the functional human gut microbiota". PLOS ONE. 6 (3): e17447. Bibcode:2011PLoSO...617447G. doi: 10.1371/journal.pone.0017447 . PMC 3050895 . PMID 21408168.
- ↑ Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, Izard J, Garrett WS, Chan AT, Huttenhower C (June 2014). "Relating the metatranscriptome and metagenome of the human gut". Proceedings of the National Academy of Sciences of the United States of America. 111 (22): E2329–38. Bibcode:2014PNAS..111E2329F. doi: 10.1073/pnas.1319284111 . PMC 4050606 . PMID 24843156.
- ↑ Burisch J, Jess T, Martinato M, Lakatos PL (May 2013). "The burden of inflammatory bowel disease in Europe". Journal of Crohn's & Colitis. 7 (4): 322–37. doi: 10.1016/j.crohns.2013.01.010 . PMID 23395397.
- 1 2 3 Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, D'Amato M, Bonfiglio F, McDonald D, Gonzalez A, McClure EE, Dunklebarger MF, Knight R, Jansson JK (February 2017). "Dynamics of the human gut microbiome in inflammatory bowel disease". Nature Microbiology. 2 (5): 17004. doi:10.1038/nmicrobiol.2017.4. PMC 5319707 . PMID 28191884.
- ↑ Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, et al. (October 2015). "Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn's Disease". Cell Host & Microbe. 18 (4): 489–500. doi:10.1016/j.chom.2015.09.008. PMC 4633303 . PMID 26468751.
- ↑ Rehman A, Lepage P, Nolte A, Hellmig S, Schreiber S, Ott SJ (September 2010). "Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients". Journal of Medical Microbiology. 59 (Pt 9): 1114–22. doi: 10.1099/jmm.0.021170-0 . PMID 20522625.
- ↑ Naughton J, Duggan G, Bourke B, Clyne M (2014). "Interaction of microbes with mucus and mucins: recent developments". Gut Microbes. 5 (1): 48–52. doi:10.4161/gmic.26680. PMC 4049936 . PMID 24149677.
- ↑ Westermann AJ, Gorski SA, Vogel J (September 2012). "Dual RNA-seq of pathogen and host". Nature Reviews Microbiology. 10 (9): 618–630. doi:10.1038/nrmicro2852. PMID 22890146. S2CID 205498287.
- ↑ Saliba AE, C Santos S, Vogel J (February 2017). "New RNA-seq approaches for the study of bacterial pathogens". Current Opinion in Microbiology. 35: 78–87. doi:10.1016/j.mib.2017.01.001. hdl: 10033/621506 . PMID 28214646.