Related Research Articles

A bone tumor is an abnormal growth of tissue in bone, traditionally classified as noncancerous (benign) or cancerous (malignant). Cancerous bone tumors usually originate from a cancer in another part of the body such as from lung, breast, thyroid, kidney and prostate. There may be a lump, pain, or neurological signs from pressure. A bone tumor might present with a pathologic fracture. Other symptoms may include fatigue, fever, weight loss, anemia and nausea. Sometimes there are no symptoms and the tumour is found when investigating another problem.
Rhabdomyosarcoma (RMS) is a highly aggressive form of cancer that develops from mesenchymal cells that have failed to fully differentiate into myocytes of skeletal muscle. Cells of the tumor are identified as rhabdomyoblasts.

Dermatofibrosarcoma protuberans (DFSP) is a rare locally aggressive malignant cutaneous soft-tissue sarcoma. DFSP develops in the connective tissue cells in the middle layer of the skin (dermis). Estimates of the overall occurrence of DFSP in the United States are 0.8 to 4.5 cases per million persons per year. In the United States, DFSP accounts for between 1 and 6 percent of all soft tissue sarcomas and 18 percent of all cutaneous soft tissue sarcomas. In the Surveillance, Epidemiology and End Results (SEER) tumor registry from 1992 through 2004, DFSP was second only to Kaposi sarcoma.

Chondrosarcoma is a bone sarcoma, a primary cancer composed of cells derived from transformed cells that produce cartilage. A chondrosarcoma is a member of a category of tumors of bone and soft tissue known as sarcomas. About 30% of bone sarcomas are chondrosarcomas. It is resistant to chemotherapy and radiotherapy. Unlike other primary bone sarcomas that mainly affect children and adolescents, a chondrosarcoma can present at any age. It more often affects the axial skeleton than the appendicular skeleton.
A blastoma is a type of cancer, more common in children, that is caused by malignancies in precursor cells, often called blasts. Examples are nephroblastoma, medulloblastoma, and retinoblastoma. The suffix -blastoma is used to imply a tumor of primitive, incompletely differentiated cells, e.g., chondroblastoma is composed of cells resembling the precursor of chondrocytes.

Primitive neuroectodermal tumor is a malignant (cancerous) neural crest tumor. It is a rare tumor, usually occurring in children and young adults under 25 years of age. The overall 5 year survival rate is about 53%.

Primary tumors of the heart are extremely rare tumors that arise from the normal tissues that make up the heart. The incidence of primary cardiac tumors has been found to be approximately 0.02%. This is in contrast to secondary tumors of the heart, which are typically either metastatic from another part of the body, or infiltrate the heart via direct extension from the surrounding tissues. Metastatic tumors to the heart are about 20 times more common than primary cardiac tumors.
Vaginal cancer is an extraordinarily rare form of cancer that develops in the tissue of the vagina. Primary vaginal cancer originates from the vaginal tissue – most frequently squamous cell carcinoma, but primary vaginal adenocarcinoma, sarcoma, and melanoma have also been reported – while secondary vaginal cancer involves the metastasis of a cancer that originated in a different part of the body. Secondary vaginal cancer is more common. Signs of vaginal cancer may include abnormal vaginal bleeding, dysuria, tenesmus, or pelvic pain, though as many as 20% of women diagnosed with vaginal cancer are asymptomatic at the time of diagnosis. Vaginal cancer occurs more frequently in women over age 50, and the mean age of diagnosis of vaginal cancer is 60 years. It often can be cured if found and treated in early stages. Surgery alone or surgery combined with pelvic radiation is typically used to treat vaginal cancer.

Congenital mesoblastic nephroma, while rare, is the most common kidney neoplasm diagnosed in the first three months of life and accounts for 3-5% of all childhood renal neoplasms. This neoplasm is generally non-aggressive and amenable to surgical removal. However, a readily identifiable subset of these kidney tumors has a more malignant potential and is capable of causing life-threatening metastases. Congenital mesoblastic nephroma was first named as such in 1967 but was recognized decades before this as fetal renal hamartoma or leiomyomatous renal hamartoma.

Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid-like features. It accounts for less than 1% of all soft tissue sarcomas. It was first clearly characterized by F.M. Enzinger in 1970. It commonly presents itself in the distal limbs of young adults as a small, soft mass or a series of bumps. A proximal version has also been described, frequently occurring in the upper extremities. Rare cases have been reported in the pelvis, vulva, penis, and spine.

Epithelioid hemangioendothelioma (eHAE) is a rare tumor, first characterized by Sharon Weiss and Franz Enzinger in 1982 that both clinically and histologically is intermediate between angiosarcoma and hemangioma. However, a distinct, disease-defining genetic alteration recently described for EHE indicates that it is an entirely separate entity from both angiosarcoma and hemangioma.

Bone metastasis, or osseous metastatic disease, is a category of cancer metastases that results from primary tumor invasion to bone. Bone-originating primary tumors such as osteosarcoma, chondrosarcoma, and Ewing's sarcoma are rare; the most common bone tumor is a metastasis. Bone metastases can be classified as osteolytic, osteoblastic, or both. Unlike hematologic malignancies which originate in the blood and form non-solid tumors, bone metastases generally arise from epithelial tumors and form a solid mass inside the bone. Bone metastases, especially in a state of advanced disease, can cause severe pain, characterized by a dull, constant ache with periodic spikes of incident pain.

Combined small cell lung carcinoma is a form of multiphasic lung cancer that is diagnosed by a pathologist when a malignant tumor, arising from transformed cells originating in lung tissue, contains a component of;small cell lung carcinoma (SCLC), admixed with one components of any histological variant of non-small cell lung carcinoma (NSCLC) in any relative proportion.

Fetal adenocarcinoma (FA) of the lung is a rare subtype of pulmonary adenocarcinoma that exhibits tissue architecture and cell characteristics that resemble fetal lung tissue upon microscopic examination. It is currently considered a variant of solid adenocarcinoma with mucin production.
Large cell lung carcinoma with rhabdoid phenotype (LCLC-RP) is a rare histological form of lung cancer, currently classified as a variant of large cell lung carcinoma (LCLC). In order for a LCLC to be subclassified as the rhabdoid phenotype variant, at least 10% of the malignant tumor cells must contain distinctive structures composed of tangled intermediate filaments that displace the cell nucleus outward toward the cell membrane. The whorled eosinophilic inclusions in LCLC-RP cells give it a microscopic resemblance to malignant cells found in rhabdomyosarcoma (RMS), a rare neoplasm arising from transformed skeletal muscle. Despite their microscopic similarities, LCLC-RP is not associated with rhabdomyosarcoma.
Sarcomatoid carcinoma of the lung is a term that encompasses five distinct histological subtypes of lung cancer, including (1) pleomorphic carcinoma, (2) spindle cell carcinoma, (3) giant cell carcinoma, (4) carcinosarcoma, or (5) pulmonary blastoma.

Giant-cell carcinoma of the lung (GCCL) is a rare histological form of large-cell lung carcinoma, a subtype of undifferentiated lung cancer, traditionally classified within the non-small-cell lung carcinomas (NSCLC).
A rhabdomyoblast is a cell type which is found in some rhabdomyosarcomas. When found histologically, a rhabdomyoblast aids the diagnosis of embryonal, alveolar, spindle cell/sclerosing, and pleomorphic rhabdomyosarcomas; however, in a tumor, expression of the rhabdomyoblast phenotype is not the only factor in diagnosing a rhabdomyosarcoma. Mesenchymal malignancies can exhibit this phenotype as well. Immunohistochemistry techniques allow for the sensitive detection of desmin, vimentin, muscle specific actin, and MyoD1. Similarly the rhabdomyoblast phenotype can be detected morphologically. Rhabdomyoblasts are early stage mesenchymal cells, having the potential to differentiate into a wide range of skeletal cells. Each stage of differentiation exhibits unique and distinguishable histological characteristics. In its initial from, stellate cells with amphiphilic cytoplasm and ovular central nuclei are observed. Commonly referred to as rhabdoid features, the maturing rhabdomyoblast will likely exhibit low levels of eosinophilic cytoplasm in proximal distances to the nucleus. As maturation and differentiation progress, the cell's cytoplasmic levels of white blood cells increase; additionally, elongated shapes, commonly depicted as “tadpole”, “strap” and "spider cells", are observed. In the concluding phase of differentiation, the white blood cell rich cytoplasm appears bright and exhibits cross-striation. The highly regulated organization of actin and myosin microfilaments in contractile proteins results in this appearance.
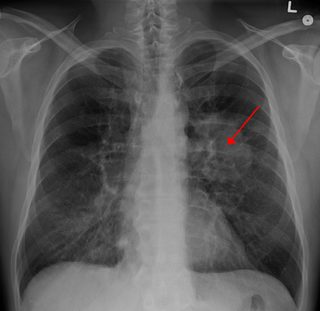
Lung tumors are neoplastic lung nodules. These include:
Cancer in adolescents and young adults is cancer which occurs in those between the ages of 15 and 39. This occurs in about 70,000 people a year in the United States—accounting for about 5 percent of cancers. This is about six times the number of cancers diagnosed in children ages 0–14. Globally, nearly 1 million young adults between the ages of 20 and 39 were diagnosed with cancer in 2012, and more than 350,000 people in this age range died from cancer.
References
- 1 2 Dishop MK, Kuruvilla S (July 2008). "Primary and metastatic lung tumors in the pediatric population: a review and 25-year experience at a large children's hospital". Arch. Pathol. Lab. Med. 132 (7): 1079–103. doi:10.5858/2008-132-1079-PAMLTI. PMID 18605764.
- ↑ Indolfi P, Casale F, Carli M, et al. (September 2000). "Pleuropulmonary blastoma: management and prognosis of 11 cases". Cancer. 89 (6): 1396–401. doi: 10.1002/1097-0142(20000915)89:6<1396::AID-CNCR25>3.0.CO;2-2 . PMID 11002236.
- ↑ Cakir O, Topal U, Bayram AS, Tolunay S (March 2005). "Sarcomas: rare primary malignant tumors of the thorax". Diagn Interv Radiol. 11 (1): 23–7. PMID 15795839.
- ↑ Cai S, Wang X, Zhao W, Fu L, Ma X, Peng X (2017) DICER1 mutations in twelve Chinese patients with pleuropulmonary blastoma. Sci China Life Sci doi: 10.1007/s11427-017-9081-x
- 1 2 Hill DA, Jarzembowski JA, Priest JR, Williams G, Schoettler P, Dehner LP (February 2008). "Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the international pleuropulmonary blastoma registry". Am. J. Surg. Pathol. 32 (2): 282–95. doi:10.1097/PAS.0b013e3181484165. PMID 18223332. S2CID 3193037.
- ↑ Priest JR, Magnuson J, Williams GM, Abromowitch M, Byrd R, Sprinz P, Finkelstein M, Moertel CL, Hill DA (September 2007). "Cerebral metastasis and other central nervous system complications of pleuropulmonary blastoma". Pediatr Blood Cancer. 49 (3): 266–73. doi:10.1002/pbc.20937. PMID 16807914. S2CID 20486633.
- ↑ Manivel JC, Priest JR, Watterson J, et al. (October 1988). "Pleuropulmonary blastoma. The so-called pulmonary blastoma of childhood". Cancer. 62 (8): 1516–26. doi: 10.1002/1097-0142(19881015)62:8<1516::AID-CNCR2820620812>3.0.CO;2-3 . PMID 3048630.