
A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant (cancerous) tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.

A soft-tissue sarcoma (STS) is a malignant tumor, a type of cancer, that develops in soft tissue. A soft-tissue sarcoma is often a painless mass that grows slowly over months or years. They may be superficial or deep-seated. Any such unexplained mass must be diagnosed by biopsy. Treatment may include surgery, radiotherapy, chemotherapy, and targeted drug therapy. Bone sarcomas are the other class of sarcomas.

A bone tumor is an abnormal growth of tissue in bone, traditionally classified as noncancerous (benign) or cancerous (malignant). Cancerous bone tumors usually originate from a cancer in another part of the body such as from lung, breast, thyroid, kidney and prostate. There may be a lump, pain, or neurological signs from pressure. A bone tumor might present with a pathologic fracture. Other symptoms may include fatigue, fever, weight loss, anemia and nausea. Sometimes there are no symptoms and the tumour is found when investigating another problem.

An osteosarcoma (OS) or osteogenic sarcoma (OGS) is a cancerous tumor in a bone. Specifically, it is an aggressive malignant neoplasm that arises from primitive transformed cells of mesenchymal origin and that exhibits osteoblastic differentiation and produces malignant osteoid.
Rhabdomyosarcoma (RMS) is a highly aggressive form of cancer that develops from mesenchymal cells that have failed to fully differentiate into myocytes of skeletal muscle. Cells of the tumor are identified as rhabdomyoblasts.

Liposarcomas are the most common subtype of soft tissue sarcomas, accounting for at least 20% of all sarcomas in adults. Soft tissue sarcomas are rare neoplasms with over 150 different histological subtypes or forms. Liposarcomas arise from the precursor lipoblasts of the adipocytes in adipose tissues. Adipose tissues are distributed throughout the body, including such sites as the deep and more superficial layers of subcutaneous tissues as well as in less surgically accessible sites like the retroperitoneum and visceral fat inside the abdominal cavity.

A malignant peripheral nerve sheath tumor (MPNST) is a form of cancer of the connective tissue surrounding nerves. Given its origin and behavior it is classified as a sarcoma. About half the cases are diagnosed in people with neurofibromatosis; the lifetime risk for an MPNST in patients with neurofibromatosis type 1 is 8–13%. MPNST with rhabdomyoblastomatous component are called malignant triton tumors.
The International Classification of Diseases for Oncology (ICD-O) is a domain-specific extension of the International Statistical Classification of Diseases and Related Health Problems for tumor diseases. This classification is widely used by cancer registries.
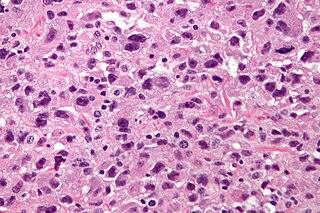
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO), 2020, as a rare, poorly differentiated neoplasm, i.e. an abnormal growth of cells that have an unclear identity and/or cell of origin. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers known or thought to derive from mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.

Ewing sarcoma is a type of pediatric cancer that forms in bone or soft tissue. Symptoms may include swelling and pain at the site of the tumor, fever, and a bone fracture. The most common areas where it begins are the legs, pelvis, and chest wall. In about 25% of cases, the cancer has already spread to other parts of the body at the time of diagnosis. Complications may include a pleural effusion or paraplegia.
Soft tissue sarcoma refers to a broad group of tumors that originate from connective tissues. They tend to have similar histologic appearance and biological behavior, and can be either benign or malignant. Soft tissue sarcomas can arise in any part of the pet's body but skin and subcutaneous tumors are the most commonly observed. Soft-tissue sarcomas comprise approximately 15% of all skin and subcutaneous tumors in dogs and approximately 7% of all skin and subcutaneous tumors in cats. The variety of different tumors that fall under the category of soft tissue sarcomas includes fibrosarcoma, hemangiopericytoma, liposarcoma, rhabdomyosarcoma, leiomyosarcoma, malignant fibrous histiocytoma, malignant nerve sheath tumors, myxosarcoma, myxofibrosarcoma, mesenchymoma, and spindle cell tumor.

Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid-like features. It accounts for less than 1% of all soft tissue sarcomas. It was first definitively characterized by F.M. Enzinger in 1970. It commonly presents itself in the distal limbs of young adults as a small, soft mass or a cluster of bumps. A proximal version has also been described, frequently occurring in the upper extremities. Less commonly, cases are reported in the pelvis, vulva, penis, and spine.
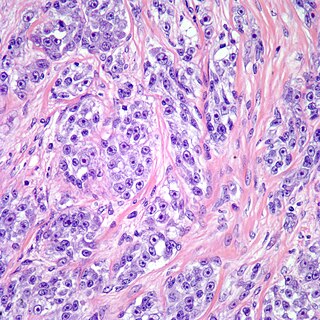
Clear cell sarcoma is a rare form of cancer called a sarcoma. It is known to occur mainly in the soft tissues and dermis. Rare forms were thought to occur in the gastrointestinal tract before they were discovered to be different and redesignated as gastrointestinal neuroectodermal tumors.
Embryonal rhabdomyosarcoma (EMRS) is a rare histological form of cancer in the connective tissue wherein the mesenchymally-derived malignant cells resemble the primitive developing skeletal muscle of the embryo. It is the most common soft tissue sarcoma occurring in children. Embryonal rhabdomyosarcoma is also known as PAX-fusion negative or fusion-negative rhabdomyosarcoma, as tumors of this subtype are unified by their lack of a PAX3-FOXO1 fusion oncogene. Fusion status refers to the presence or absence of a fusion gene, which is a gene formed from joining two different genes together through DNA rearrangements. These types of tumors are classified as embryonal rhabdomyosarcoma "because of their remarkable resemblance to developing embryonic and fetal skeletal muscle."

Low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade sarcoma first described by H. L. Evans in 1987. LGFMS are soft tissue tumors of the mesenchyme-derived connective tissues; on microscopic examination, they are found to be composed of spindle-shaped cells that resemble fibroblasts. These fibroblastic, spindle-shaped cells are neoplastic cells that in most cases of LGFMS express fusion genes, i.e. genes composed of parts of two different genes that form as a result of mutations. The World Health Organization (2020) classified LGFMS as a specific type of tumor in the category of malignant fibroblastic and myofibroblastic tumors.
A rhabdomyoblast is a cell type which is found in some rhabdomyosarcomas. When found histologically, a rhabdomyoblast aids the diagnosis of embryonal, alveolar, spindle cell/sclerosing, and pleomorphic rhabdomyosarcomas; however, in a tumor, expression of the rhabdomyoblast phenotype is not the only factor in diagnosing a rhabdomyosarcoma. Mesenchymal malignancies can exhibit this phenotype as well. Immunohistochemistry techniques allow for the sensitive detection of desmin, vimentin, muscle specific actin, and MyoD1. Similarly the rhabdomyoblast phenotype can be detected morphologically. Rhabdomyoblasts are early stage mesenchymal cells, having the potential to differentiate into a wide range of skeletal cells. Each stage of differentiation exhibits unique and distinguishable histological characteristics. In its initial from, stellate cells with amphiphilic cytoplasm and ovular central nuclei are observed. Commonly referred to as rhabdoid features, the maturing rhabdomyoblast will likely exhibit low levels of eosinophilic cytoplasm in proximal distances to the nucleus. As maturation and differentiation progress, the cell's cytoplasmic levels of white blood cells increase; additionally, elongated shapes, commonly depicted as “tadpole”, “strap” and "spider cells", are observed. In the concluding phase of differentiation, the white blood cell rich cytoplasm appears bright and exhibits cross-striation. The highly regulated organization of actin and myosin microfilaments in contractile proteins results in this appearance.
Myxofibrosarcoma (MFS), although a rare type of tumor, is one of the most common soft tissue sarcomas, i.e. cancerous tumors, that develop in the soft tissues of elderly individuals. Initially considered to be a type of histiocytoma termed fibrous histiocytoma or myxoid variant of malignant fibrous histiocytoma, Angervall et al. termed this tumor myxofibrosarcoma in 1977. In 2020, the World Health Organization reclassified MFS as a separate and distinct tumor in the category of malignant fibroblastic and myofibroblastic tumors.
Sclerosing epithelioid fibrosarcoma (SEF) is a very rare malignant tumor of soft tissues that on microscopic examination consists of small round or ovoid neoplastic epithelioid fibroblast-like cells, i.e. cells that have features resembling both epithelioid cells and fibroblasts. In 2020, the World Health Organization classified SEF as a distinct tumor type in the category of malignant fibroblastic and myofibroblastic tumors. However, current studies have reported that low-grade fibromyxoid sarcoma (LGFMS) has many clinically and pathologically important features characteristic of SEF; these studies suggest that LGSFMS may be an early form of, and over time progress to become, a SEF. Since the World Health Organization has classified LGFMS as one of the malignant fibroblastic and myofibroblastic tumors that is distinctly different than SEF, SEF and LGFMS are here regarded as different tumor forms.
Low-grade myofibroblastic sarcoma (LGMS) is a subtype of the malignant sarcomas. As it is currently recognized, LGMS was first described as a rare, atypical myofibroblastic tumor by Mentzel et al. in 1998. Myofibroblastic sarcomas had been divided into low-grade myofibroblastic sarcomas, intermediate‐grade myofibroblasic sarcomas, i.e. IGMS, and high‐grade myofibroblasic sarcomas, i.e. HGMS based on their microscopic morphological, immunophenotypic, and malignancy features. LGMS and IGMS are now classified together by the World Health Organization (WHO), 2020, in the category of intermediate fibroblastic and myofibroblastic tumors. WHO, 2020, classifies HGMS as a soft tissue tumor in the category of tumors of uncertain differentiation. This article follows the WHO classification: here, LGMS includes IGMS but not HGMS which is a more aggressive and metastasizing tumor than LGMS and consists of cells of uncertain origin.
