
Saccharomyces cerevisiae is a species of yeast. The species has been instrumental in winemaking, baking, and brewing since ancient times. It is believed to have been originally isolated from the skin of grapes. It is one of the most intensively studied eukaryotic model organisms in molecular and cell biology, much like Escherichia coli as the model bacterium. It is the microorganism behind the most common type of fermentation. S. cerevisiae cells are round to ovoid, 5–10 μm in diameter. It reproduces by budding.

Yeast artificial chromosomes (YACs) are genetically engineered chromosomes derived from the DNA of the yeast, Saccharomyces cerevisiae, which is then ligated into a bacterial plasmid. By inserting large fragments of DNA, from 100–1000 kb, the inserted sequences can be cloned and physically mapped using a process called chromosome walking. This is the process that was initially used for the Human Genome Project, however due to stability issues, YACs were abandoned for the use of Bacterial artificial chromosomes (BAC). Beginning with the initial research of the Rankin et al., Strul et al., and Hsaio et al., the inherently fragile chromosome was stabilized by discovering the necessary autonomously replicating sequence (ARS); a refined YAC utilizing this data was described in 1983 by Murray et al.
A protein complex or multiprotein complex is a group of two or more associated polypeptide chains. Protein complexes are distinct from multienzyme complexes, in which multiple catalytic domains are found in a single polypeptide chain.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "gene-by-gene" approach.
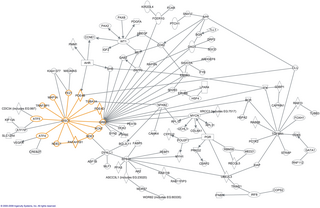
In molecular biology, an interactome is the whole set of molecular interactions in a particular cell. The term specifically refers to physical interactions among molecules but can also describe sets of indirect interactions among genes.

Two-hybrid screening is a molecular biology technique used to discover protein–protein interactions (PPIs) and protein–DNA interactions by testing for physical interactions between two proteins or a single protein and a DNA molecule, respectively.

Protein–protein interactions (PPIs) are physical contacts of high specificity established between two or more protein molecules as a result of biochemical events steered by interactions that include electrostatic forces, hydrogen bonding and the hydrophobic effect. Many are physical contacts with molecular associations between chains that occur in a cell or in a living organism in a specific biomolecular context.
petite (ρ–) is a mutant first discovered in the yeast Saccharomyces cerevisiae. Due to the defect in the respiratory chain, 'petite' yeast are unable to grow on media containing only non-fermentable carbon sources and form small colonies when grown in the presence of fermentable carbon sources. The petite phenotype can be caused by the absence of, or mutations in, mitochondrial DNA, or by mutations in nuclear-encoded genes involved in oxidative phosphorylation. A neutral petite produces all wild type progeny when crossed with wild type.
Recombineering is a genetic and molecular biology technique based on homologous recombination systems, as opposed to the older/more common method of using restriction enzymes and ligases to combine DNA sequences in a specified order. Recombineering is widely used for bacterial genetics, in the generation of target vectors for making a conditional mouse knockout, and for modifying DNA of any source often contained on a bacterial artificial chromosome (BAC), among other applications.
The Saccharomyces Genome Database (SGD) is a scientific database of the molecular biology and genetics of the yeast Saccharomyces cerevisiae, which is commonly known as baker's or budding yeast.

Oncogenomics is a sub-field of genomics that characterizes cancer-associated genes. It focuses on genomic, epigenomic and transcript alterations in cancer.

DNA repair and recombination protein RAD54-like is a protein that in humans is encoded by the RAD54L gene.
Synthetic lethality is defined as a type of genetic interaction where the combination of two genetic events results in cell death or death of an organism. Although the foregoing explanation is wider than this, it is common when referring to synthetic lethality to mean the situation arising by virtue of a combination of deficiencies of two or more genes leading to cell death, whereas a deficiency of only one of these genes does not. In a synthetic lethal genetic screen, it is necessary to begin with a mutation that does not result in cell death, although the effect of that mutation could result in a differing phenotype, and then systematically test other mutations at additional loci to determine which, in combination with the first mutation, causes cell death arising by way of deficiency or abolition of expression.
Epistasis refers to genetic interactions in which the mutation of one gene masks the phenotypic effects of a mutation at another locus. Systematic analysis of these epistatic interactions can provide insight into the structure and function of genetic pathways. Examining the phenotypes resulting from pairs of mutations helps in understanding how the function of these genes intersects. Genetic interactions are generally classified as either Positive/Alleviating or Negative/Aggravating. Fitness epistasis is positive when a loss of function mutation of two given genes results in exceeding the fitness predicted from individual effects of deleterious mutations, and it is negative when it decreases fitness. Ryszard Korona and Lukas Jasnos showed that the epistatic effect is usually positive in Saccharomyces cerevisiae. Usually, even in case of positive interactions double mutant has smaller fitness than single mutants. The positive interactions occur often when both genes lie within the same pathway Conversely, negative interactions are characterized by an even stronger defect than would be expected in the case of two single mutations, and in the most extreme cases the double mutation is lethal. This aggravated phenotype arises when genes in compensatory pathways are both knocked out.
Disease gene identification is a process by which scientists identify the mutant genotypes responsible for an inherited genetic disorder. Mutations in these genes can include single nucleotide substitutions, single nucleotide additions/deletions, deletion of the entire gene, and other genetic abnormalities.
The yeast deletion project, formally the Saccharomyces Genome Deletion Project, is a project to create data for a near-complete collection of gene-deletion mutants of the yeast Saccharomyces cerevisiae. Each strain carries a precise deletion of one of the genes in the genome. This allows researchers to determine what each gene does by comparing the mutated yeast to the behavior of normal yeast. Gene deletion, or gene knockout, is one of the main ways in which the function of genes are discovered. Many of the deletion mutations are sold by the biotech firm Invitrogen.
Reverse genetics is a method in molecular genetics that is used to help understand the function(s) of a gene by analysing the phenotypic effects caused by genetically engineering specific nucleic acid sequences within the gene. The process proceeds in the opposite direction to forward genetic screens of classical genetics. While forward genetics seeks to find the genetic basis of a phenotype or trait, reverse genetics seeks to find what phenotypes are controlled by particular genetic sequences.
Essential genes are indispensable genes for organisms to grow and reproduce offspring under certain environment. However, being essential is highly dependent on the circumstances in which an organism lives. For instance, a gene required to digest starch is only essential if starch is the only source of energy. Recently, systematic attempts have been made to identify those genes that are absolutely required to maintain life, provided that all nutrients are available. Such experiments have led to the conclusion that the absolutely required number of genes for bacteria is on the order of about 250–300. Essential genes of single-celled organisms encode proteins for three basic functions including genetic information processing, cell envelopes and energy production. Those gene functions are used to maintain a central metabolism, replicate DNA, translate genes into proteins, maintain a basic cellular structure, and mediate transport processes into and out of the cell. Compared with single-celled organisms, multicellular organisms have more essential genes related to communication and development. Most of the essential genes in viruses are related to the processing and maintenance of genetic information. In contrast to most single-celled organisms, viruses lack many essential genes for metabolism, which forces them to hijack the host's metabolism. Most genes are not essential but convey selective advantages and increased fitness. Hence, the vast majority of genes are not essential and many can be deleted without consequences, at least under most circumstances.
Transposon insertion sequencing (Tn-seq) combines transposon insertional mutagenesis with massively parallel sequencing (MPS) of the transposon insertion sites to identify genes contributing to a function of interest in bacteria. The method was originally established by concurrent work in four laboratories under the acronyms HITS, INSeq, TraDIS, and Tn-Seq. Numerous variations have been subsequently developed and applied to diverse biological systems. Collectively, the methods are often termed Tn-Seq as they all involve monitoring the fitness of transposon insertion mutants via DNA sequencing approaches.
The Gal4 transcription factor is a positive regulator of gene expression of galactose-induced genes. This protein represents a large fungal family of transcription factors, Gal4 family, which includes over 50 members in the yeast Saccharomyces cerevisiae e.g. Oaf1, Pip2, Pdr1, Pdr3, Leu3.

