Related Research Articles
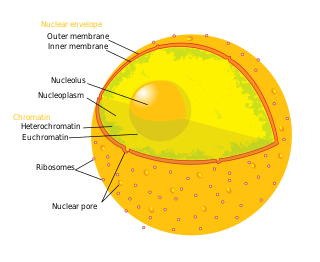
The nucleolus is the largest structure in the nucleus of eukaryotic cells. It is best known as the site of ribosome biogenesis, which is the synthesis of ribosomes. The nucleolus also participates in the formation of signal recognition particles and plays a role in the cell's response to stress. Nucleoli are made of proteins, DNA and RNA, and form around specific chromosomal regions called nucleolar organizing regions. Malfunction of nucleoli can be the cause of several human conditions called "nucleolopathies" and the nucleolus is being investigated as a target for cancer chemotherapy.

Ribosomes are macromolecular machines, found within all cells, that perform biological protein synthesis. Ribosomes link amino acids together in the order specified by the codons of messenger RNA (mRNA) molecules to form polypeptide chains. Ribosomes consist of two major components: the small and large ribosomal subunits. Each subunit consists of one or more ribosomal RNA (rRNA) molecules and many ribosomal proteins. The ribosomes and associated molecules are also known as the translational apparatus.

In biology, translation is the process in living cells in which proteins are produced using RNA molecules as templates. The generated protein is a sequence of amino acids. This sequence is determined by the sequence of nucleotides in the RNA. The nucleotides are considered three at a time. Each such triple results in addition of one specific amino acid to the protein being generated. The matching from nucleotide triple to amino acid is called the genetic code. The translation is performed by a large complex of functional RNA and proteins called ribosomes. The entire process is called gene expression.

Ribosomal ribonucleic acid (rRNA) is a type of non-coding RNA which is the primary component of ribosomes, essential to all cells. rRNA is a ribozyme which carries out protein synthesis in ribosomes. Ribosomal RNA is transcribed from ribosomal DNA (rDNA) and then bound to ribosomal proteins to form small and large ribosome subunits. rRNA is the physical and mechanical factor of the ribosome that forces transfer RNA (tRNA) and messenger RNA (mRNA) to process and translate the latter into proteins. Ribosomal RNA is the predominant form of RNA found in most cells; it makes up about 80% of cellular RNA despite never being translated into proteins itself. Ribosomes are composed of approximately 60% rRNA and 40% ribosomal proteins by mass.
An internal ribosome entry site, abbreviated IRES, is an RNA element that allows for translation initiation in a cap-independent manner, as part of the greater process of protein synthesis. In eukaryotic translation, initiation typically occurs at the 5' end of mRNA molecules, since 5' cap recognition is required for the assembly of the initiation complex. The location for IRES elements is often in the 5'UTR, but can also occur elsewhere in mRNAs.
Bacterial translation is the process by which messenger RNA is translated into proteins in bacteria.
Eukaryotic translation is the biological process by which messenger RNA is translated into proteins in eukaryotes. It consists of four phases: initiation, elongation, termination, and recapping.
The Kozak consensus sequence is a nucleic acid motif that functions as the protein translation initiation site in most eukaryotic mRNA transcripts. Regarded as the optimum sequence for initiating translation in eukaryotes, the sequence is an integral aspect of protein regulation and overall cellular health as well as having implications in human disease. It ensures that a protein is correctly translated from the genetic message, mediating ribosome assembly and translation initiation. A wrong start site can result in non-functional proteins. As it has become more studied, expansions of the nucleotide sequence, bases of importance, and notable exceptions have arisen. The sequence was named after the scientist who discovered it, Marilyn Kozak. Kozak discovered the sequence through a detailed analysis of DNA genomic sequences.
A ribosome binding site, or ribosomal binding site (RBS), is a sequence of nucleotides upstream of the start codon of an mRNA transcript that is responsible for the recruitment of a ribosome during the initiation of translation. Mostly, RBS refers to bacterial sequences, although internal ribosome entry sites (IRES) have been described in mRNAs of eukaryotic cells or viruses that infect eukaryotes. Ribosome recruitment in eukaryotes is generally mediated by the 5' cap present on eukaryotic mRNAs.

The prokaryotic small ribosomal subunit, or 30S subunit, is the smaller subunit of the 70S ribosome found in prokaryotes. It is a complex of the 16S ribosomal RNA (rRNA) and 19 proteins. This complex is implicated in the binding of transfer RNA to messenger RNA (mRNA). The small subunit is responsible for the binding and the reading of the mRNA during translation. The small subunit, both the rRNA and its proteins, complexes with the large 50S subunit to form the 70S prokaryotic ribosome in prokaryotic cells. This 70S ribosome is then used to translate mRNA into proteins.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.

EF-G is a prokaryotic elongation factor involved in protein translation. As a GTPase, EF-G catalyzes the movement (translocation) of transfer RNA (tRNA) and messenger RNA (mRNA) through the ribosome.

RNA-Seq is a sequencing technique that uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample, representing an aggregated snapshot of the cells' dynamic pool of RNAs, also known as transcriptome.
Ribosome profiling, or Ribo-Seq, is an adaptation of a technique developed by Joan Steitz and Marilyn Kozak almost 50 years ago that Nicholas Ingolia and Jonathan Weissman adapted to work with next generation sequencing that uses specialized messenger RNA (mRNA) sequencing to determine which mRNAs are being actively translated. A related technique that can also be used to determine which mRNAs are being actively translated is the Translating Ribosome Affinity Purification (TRAP) methodology, which was developed by Nathaniel Heintz at Rockefeller University. TRAP does not involve ribosome footprinting but provides cell type-specific information.

Ribosomal pause refers to the queueing or stacking of ribosomes during translation of the nucleotide sequence of mRNA transcripts. These transcripts are decoded and converted into an amino acid sequence during protein synthesis by ribosomes. Due to the pause sites of some mRNA's, there is a disturbance caused in translation. Ribosomal pausing occurs in both eukaryotes and prokaryotes. A more severe pause is known as a ribosomal stall.

Polysome profiling is a technique in molecular biology that is used to study the association of mRNAs with ribosomes. It is important to note that this technique is different from ribosome profiling. Both techniques have been reviewed and both are used in analysis of the translatome, but the data they generate are at very different levels of specificity. When employed by experts, the technique is remarkably reproducible: the 3 profiles in the first image are from 3 different experiments.

In epitranscriptomic sequencing, most methods focus on either (1) enrichment and purification of the modified RNA molecules before running on the RNA sequencer, or (2) improving or modifying bioinformatics analysis pipelines to call the modification peaks. Most methods have been adapted and optimized for mRNA molecules, except for modified bisulfite sequencing for profiling 5-methylcytidine which was optimized for tRNAs and rRNAs.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.

Translatomics is the study of all open reading frames (ORFs) that are being actively translated in a cell or organism. This collection of ORFs is called the translatome. Characterizing a cell's translatome can give insight into the array of biological pathways that are active in the cell. According to the central dogma of molecular biology, the DNA in a cell is transcribed to produce RNA, which is then translated to produce a protein. Thousands of proteins are encoded in an organism's genome, and the proteins present in a cell cooperatively carry out many functions to support the life of the cell. Under various conditions, such as during stress or specific timepoints in development, the cell may require different biological pathways to be active, and therefore require a different collection of proteins. Depending on intrinsic and environmental conditions, the collection of proteins being made at one time varies. Translatomic techniques can be used to take a "snapshot" of this collection of actively translating ORFs, which can give information about which biological pathways the cell is activating under the present conditions.
This glossary of genetics is a list of definitions of terms and concepts commonly used in the study of genetics and related disciplines in biology, including molecular biology, cell biology, and evolutionary biology. It is intended as introductory material for novices; for more specific and technical detail, see the article corresponding to each term. For related terms, see Glossary of evolutionary biology.
References
- 1 2 Archer, Stuart K.; Shirokikh, Nikolay E.; Beilharz, Traude H.; Preiss, Thomas (2016-07-20). "Dynamics of ribosome scanning and recycling revealed by translation complex profiling". Nature. 535 (7613): 570–4. Bibcode:2016Natur.535..570A. doi:10.1038/nature18647. ISSN 1476-4687. PMID 27437580. S2CID 4464952.
- ↑ Mašek, Tomáš; Valášek, Leoš; Pospíšek, Martin (2011-01-01). "Polysome Analysis and RNA Purification from Sucrose Gradients". RNA. Methods in Molecular Biology. Vol. 703. pp. 293–309. doi:10.1007/978-1-59745-248-9_20. ISBN 978-1-58829-913-0. ISSN 1940-6029. PMID 21125498.
- ↑ Spangenberg, Lucia; Shigunov, Patricia; Abud, Ana Paula R.; Cofré, Axel R.; Stimamiglio, Marco A.; Kuligovski, Crisciele; Zych, Jaiesa; Schittini, Andressa V.; Costa, Alexandre Dias Tavares (2013-09-01). "Polysome profiling shows extensive posttranscriptional regulation during human adipocyte stem cell differentiation into adipocytes". Stem Cell Research. 11 (2): 902–912. doi: 10.1016/j.scr.2013.06.002 . ISSN 1876-7753. PMID 23845413.
- ↑ Ingolia, Nicholas T.; Ghaemmaghami, Sina; Newman, John R. S.; Weissman, Jonathan S. (2009-04-10). "Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling". Science. 324 (5924): 218–223. Bibcode:2009Sci...324..218I. doi:10.1126/science.1168978. ISSN 1095-9203. PMC 2746483 . PMID 19213877.
- ↑ "TCP-seq data browser".
- ↑ "GWIPS-viz translation data browser".