In immunology, an antigen (Ag) is a molecule, moiety, foreign particulate matter, or an allergen, such as pollen, that can bind to a specific antibody or T-cell receptor. The presence of antigens in the body may trigger an immune response.
An antibody (Ab), also known as an immunoglobulin (Ig), is a large, Y-shaped protein used by the immune system to identify and neutralize foreign objects such as pathogenic bacteria and viruses. The antibody recognizes a unique molecule of the pathogen, called an antigen. Each tip of the "Y" of an antibody contains a paratope that is specific for one particular epitope on an antigen, allowing these two structures to bind together with precision. Using this binding mechanism, an antibody can tag a microbe or an infected cell for attack by other parts of the immune system, or can neutralize it directly.

A DNA vaccine is a type of vaccine that transfects a specific antigen-coding DNA sequence into the cells of an organism as a mechanism to induce an immune response.
Antigenic drift is a kind of genetic variation in viruses, arising from the accumulation of mutations in the virus genes that code for virus-surface proteins that host antibodies recognize. This results in a new strain of virus particles that is not effectively inhibited by the antibodies that prevented infection by previous strains. This makes it easier for the changed virus to spread throughout a partially immune population. Antigenic drift occurs in both influenza A and influenza B viruses.

Plasmodium falciparum is a unicellular protozoan parasite of humans, and the deadliest species of Plasmodium that causes malaria in humans. The parasite is transmitted through the bite of a female Anopheles mosquito and causes the disease's most dangerous form, falciparum malaria. It is responsible for around 50% of all malaria cases. P. falciparum is therefore regarded as the deadliest parasite in humans. It is also associated with the development of blood cancer and is classified as a Group 2A (probable) carcinogen.
Glycophorin C plays a functionally important role in maintaining erythrocyte shape and regulating membrane material properties, possibly through its interaction with protein 4.1. Moreover, it has previously been shown that membranes deficient in protein 4.1 exhibit decreased content of glycophorin C. It is also an integral membrane protein of the erythrocyte and acts as the receptor for the Plasmodium falciparum protein PfEBP-2.
Virus-like particles (VLPs) are molecules that closely resemble viruses, but are non-infectious because they contain no viral genetic material. They can be naturally occurring or synthesized through the individual expression of viral structural proteins, which can then self assemble into the virus-like structure. Combinations of structural capsid proteins from different viruses can be used to create recombinant VLPs. Both in-vivo assembly and in-vitro assembly have been successfully shown to form virus-like particles. VLPs derived from the Hepatitis B virus (HBV) and composed of the small HBV derived surface antigen (HBsAg) were described in 1968 from patient sera. VLPs have been produced from components of a wide variety of virus families including Parvoviridae, Retroviridae, Flaviviridae, Paramyxoviridae and bacteriophages. VLPs can be produced in multiple cell culture systems including bacteria, mammalian cell lines, insect cell lines, yeast and plant cells.

The adaptive immune system, also known as the acquired immune system, or specific immune system is a subsystem of the immune system that is composed of specialized, systemic cells and processes that eliminate pathogens or prevent their growth. The acquired immune system is one of the two main immunity strategies found in vertebrates.
Trypanosoma brucei is a species of parasitic kinetoplastid belonging to the genus Trypanosoma that is present in sub-Saharan Africa. Unlike other protozoan parasites that normally infect blood and tissue cells, it is exclusively extracellular and inhabits the blood plasma and body fluids. It causes deadly vector-borne diseases: African trypanosomiasis or sleeping sickness in humans, and animal trypanosomiasis or nagana in cattle and horses. It is a species complex grouped into three subspecies: T. b. brucei, T. b. gambiense and T. b. rhodesiense. The first is a parasite of non-human mammals and causes nagana, while the latter two are zoonotic infecting both humans and animals and cause African trypanosomiasis.
Subtelomeres are segments of DNA between telomeric caps and chromatin.
In academia, computational immunology is a field of science that encompasses high-throughput genomic and bioinformatics approaches to immunology. The field's main aim is to convert immunological data into computational problems, solve these problems using mathematical and computational approaches and then convert these results into immunologically meaningful interpretations.
A blocking antibody is an antibody that does not have a reaction when combined with an antigen, but prevents other antibodies from combining with that antigen. This function of blocking antibodies has had a variety of clinical and experimental uses.
Malaria vaccines are vaccines that prevent malaria, a mosquito-borne infectious disease which annually affects an estimated 247 million people worldwide and causes 619,000 deaths. The first approved vaccine for malaria is RTS,S, known by the brand name Mosquirix. As of April 2023, the vaccine has been given to 1.5 million children living in areas with moderate-to-high malaria transmission. It requires at least three doses in infants by age 2, and a fourth dose extends the protection for another 1–2 years. The vaccine reduces hospital admissions from severe malaria by around 30%.
Antigenic escape, immune escape, immune evasion or escape mutation occurs when the immune system of a host, especially of a human being, is unable to respond to an infectious agent: the host's immune system is no longer able to recognize and eliminate a pathogen, such as a virus. This process can occur in a number of different ways of both a genetic and an environmental nature. Such mechanisms include homologous recombination, and manipulation and resistance of the host's immune responses.
A neutralizing antibody (NAb) is an antibody that defends a cell from a pathogen or infectious particle by neutralizing any effect it has biologically. Neutralization renders the particle no longer infectious or pathogenic. Neutralizing antibodies are part of the humoral response of the adaptive immune system against viruses, intracellular bacteria and microbial toxin. By binding specifically to surface structures (antigen) on an infectious particle, neutralizing antibodies prevent the particle from interacting with its host cells it might infect and destroy.
Human genetic resistance to malaria refers to inherited changes in the DNA of humans which increase resistance to malaria and result in increased survival of individuals with those genetic changes. The existence of these genotypes is likely due to evolutionary pressure exerted by parasites of the genus Plasmodium which cause malaria. Since malaria infects red blood cells, these genetic changes are most common alterations to molecules essential for red blood cell function, such as hemoglobin or other cellular proteins or enzymes of red blood cells. These alterations generally protect red blood cells from invasion by Plasmodium parasites or replication of parasites within the red blood cell.

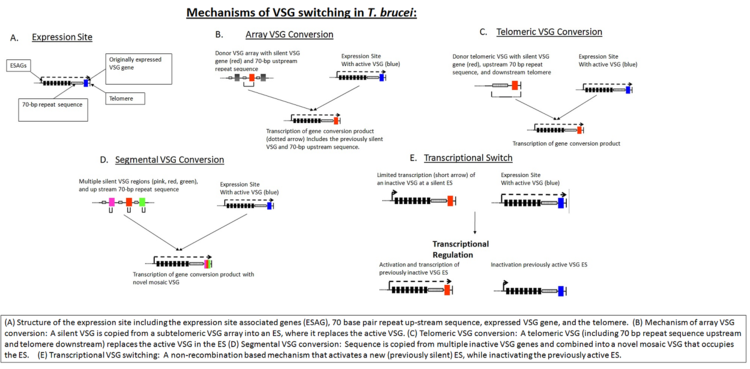
Variant surface glycoprotein (VSG) is a ~60kDa protein which densely packs the cell surface of protozoan parasites belonging to the genus Trypanosoma. This genus is notable for their cell surface proteins. They were first isolated from Trypanosoma brucei in 1975 by George Cross. VSG allows the trypanosomatid parasites to evade the mammalian host's immune system by extensive antigenic variation. They form a 12–15 nm surface coat. VSG dimers make up ~90% of all cell surface protein and ~10% of total cell protein. For this reason, these proteins are highly immunogenic and an immune response raised against a specific VSG coat will rapidly kill trypanosomes expressing this variant. However, with each cell division there is a possibility that the progeny will switch expression to change the VSG that is being expressed. VSG has no prescribed biochemical activity.
Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) is a family of proteins present on the membrane surface of red blood cells that are infected by the malarial parasite Plasmodium falciparum. PfEMP1 is synthesized during the parasite's blood stage inside the RBC, during which the clinical symptoms of falciparum malaria are manifested. Acting as both an antigen and adhesion protein, it is thought to play a key role in the high level of virulence associated with P. falciparum. It was discovered in 1984 when it was reported that infected RBCs had unusually large-sized cell membrane proteins, and these proteins had antibody-binding (antigenic) properties. An elusive protein, its chemical structure and molecular properties were revealed only after a decade, in 1995. It is now established that there is not one but a large family of PfEMP1 proteins, genetically regulated (encoded) by a group of about 60 genes called var. Each P. falciparum is able to switch on and off specific var genes to produce a functionally different protein, thereby evading the host's immune system. RBCs carrying PfEMP1 on their surface stick to endothelial cells, which facilitates further binding with uninfected RBCs, ultimately helping the parasite to both spread to other RBCs as well as bringing about the fatal symptoms of P. falciparum malaria.

The Plasmodium helical interspersed subtelomeric proteins (PHIST) or ring-infected erythrocyte surface antigens (RESA) are a family of protein domains found in the malaria-causing Plasmodium species. It was initially identified as a short four-helical conserved region in the single-domain export proteins, but the identification of this part associated with a DnaJ domain in P. falciparum RESA has led to its reclassification as the RESA N-terminal domain. This domain has been classified into three subfamilies, PHISTa, PHISTb, and PHISTc.
Bacteriophage AP205 is a plaque-forming bacteriophage that infects Acinetobacter bacteria. Bacteriophage AP205 is a protein-coated virus with a positive single-stranded RNA genome. It is a member of the family Fiersviridae, consisting of particles that infect Gram-negative bacteria such as E. coli.