Acquired resistance
Since cancer is a genetic disease, [5] two genomic events underlie these mechanisms of acquired drug resistance: Genome alterations (e.g. gene amplification and deletion) and epigenetic modifications.
Genetic causes
Genome alterations
Chromosomal rearrangement due to genome instability can cause gene amplification and deletion. Gene amplification is the increase in copy number of a region of a chromosome. [6] which occur frequently in solid tumors, and can contribute to tumor evolution through altered gene expression. [6]
Hamster cell research in 1993 showed that amplifications in the DHFR gene involved in DNA synthesis began with chromosome break in below the gene, and subsequent cycles of bridge-breakage-fusion formations result in large intrachromosomal repeats. [7] The over amplification of oncogenes can occur in response to chemotherapy, thought to be the underlying mechanism in several classes of resistance. [6] For example, DHFR amplification occurs in response to methotrexate, [8] TYMS (involved in DNA synthesis) amplification occurs in response to 5-fluorouracil, [9] and BCR-ABL amplification occurs in response to imatinib mesylate. [10] Determining areas of gene amplification in cells from cancer patients has huge clinical implications. Gene deletion is the opposite of gene amplification, where a region of a chromosome is lost and drug resistance occurs by losing tumor suppressor genes such as TP53. [2]
Genomic instability can occur when the replication fork is disturbed or stalled in its migration. This can occur with replication fork barriers, proteins such as PTIP, CHD4 and PARP1, which are normally cleared by the cell's DNA damage sensors, surveyors, and responders BRCA1 and BRCA2. [11]
Epigenetic mechanisms
Epigenetic modifications in antineoplastic drug resistance play a major role in cancer development and drug resistance as they contribute to the regulation of gene expression. [12] Two main types of epigenetic control are DNA methylation and histone methylation/acetylation. DNA methylation is the process of adding methyl groups to DNA, usually in the upstream promoter regions, which stops DNA transcription at the region and effectively silences individual genes. Histone modifications, such as deacetylation, alters chromatin formation and silence large chromosomal regions. In cancer cells, where normal regulation of gene expression breaks down, the oncogenes are activated via hypomethylation and tumor suppressors are silenced via hypermethylation. Similarly, in drug resistance development, it has been suggested that epigenetic modifications can result in the activation and overexpression of pro-drug resistance genes. [12]
Studies on cancer cell lines have shown that hypomethylation (loss of methylation) of the MDR1 gene promoter caused overexpression and the multidrug resistance. [13]
In a methotrexate resistant breast cancer cell lines without drug uptake and folate carrier expression, giving DAC, a DNA methylation inhibitor, improved drug uptake and folate carrier expression. [14]
Acquired resistance to the alkylating drug fotemustine in melanoma cell showed high MGMT activity related to the hypermethylation of the MGMT gene exons. [15]
In Imatinib resistant cell lines, silencing of the SOCS-3 gene via methylation has been shown to cause STAT3 protein activation, which caused uncontrolled proliferation. [16]
Cancer cell mechanisms
Cancer cells can become resistant to multiple drugs by altered membrane transport, enhanced DNA repair, apoptotic pathway defects, alteration of target molecules, protein and pathway mechanisms, such as enzymatic deactivation. [12]
Altered membrane transport

Many classes of antineoplastic drugs act on intracellular components and pathways, like DNA, nuclear components, meaning that they need to enter the cancer cells. The p-glycoprotein (P-gp), or the multiple drug resistance protein, is a phosphorylated and glycosylated membrane transporter that can shuttle drugs out of the cell, thereby decreasing or ablating drug efficacy. This transporter protein is encoded by the MDR1 gene and is also called the ATP-binding cassette (ABC) protein. MDR1 has promiscuous substrate specificity, allowing it to transport many structurally diverse compounds across the cell membrane, mainly hydrophobic compounds. Studies have found that the MDR1 gene can be activated and overexpressed in response to pharmaceutical drugs, thus forming the basis for resistance to many drugs. [2] Overexpression of the MDR1 gene in cancer cells is used to keep intracellular levels of antineoplastic drugs below cell-killing levels.[ citation needed ]
For example, the antibiotic rifampicin has been found to induce MDR1 expression. Experiments in different drug resistant cell lines and patient DNA revealed gene rearrangements which had initiated the activation or overexpression of MDR1. [17] A C3435T polymorphism in exon 226 of MDR1 has also been strongly correlated with p-glycoprotein activities. [18]
MDR1 is activated through NF-κB, a protein complex which acts as a transcription factor. [19] [20] [21] [22] In the rat, an NF-κB binding site is adjacent to the mdr1b gene, [23] NF-κB can be active in tumour cells because its mutated NF-κB gene or its inhibitory IκB gene mutated under chemotherapy. In colorectal cancer cells, inhibition of NF-κB or MDR1 caused increased apoptosis in response to a chemotherapeutic agent. [19]
Enhanced DNA repair
Enhanced DNA repair plays an important role in the ability for cancer cells to overcome drug-induced DNA damages.
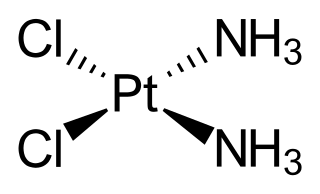
Platinum-based chemotherapies, such as cisplatin, target tumour cells by cross-linking their DNA strands, causing mutation and damage. [2] Such damage will trigger programmed cell death (e.g. apoptosis) in cancer cells. Cisplatin resistance occurs when cancer cells develop an enhanced ability to reverse such damage by removing the cisplatin from DNA and repairing any damage done. [2] [12] The cisplatin-resistant cells upregulate expression of the excision repair cross-complementing (ERCC1) gene and protein. [2]
Some chemotherapies are alkylating agents meaning they attach an alkyl group to DNA to stop it from being read. O6-methylguanine DNA methyltransferase (MGMT) is a DNA repair enzyme which removes alkyl groups from DNA. MGMT expression is upregulated in many cancer cells, which protects them from alkylating agents. [12] Increased MGMT expression has been found in colon cancer, lung cancer, non-Hodgkin's lymphoma, breast cancer, gliomas, myeloma and pancreatic cancer. [24]
Apoptotic pathway defects
TP53 is a tumor suppressor gene encoding the p53 protein, which responds to DNA damage either by DNA repair, cell cycle arrest, or apoptosis. Losing TP53 via gene deletion can allow cells to continuously replicate despite DNA damage. The tolerance of DNA damage can grant cancer cells a method of resistance to those drugs which normally induce apoptosis through DNA damage. [2] [12]
Other genes involved in the apoptotic pathway related drug resistance include h-ras and bcl-2/bax. [25] Oncogenic h-ras has been found to increase expression of ERCC1, resulting in enhanced DNA repair (see above). [26] Inhibition of h-ras was found to increase cisplatin sensitivity in glioblastoma cells. [27] Upregulated expression of Bcl-2 in leukemic cells (non-Hodgkin's lymphoma) resulted in decreased levels of apoptosis in response to chemotherapeutic agents, as Bcl-2 is a pro-survival oncogene. [28]
Altered target molecules
During targeted therapy, oftentimes the target has modified itself and decreased its expression to the point that therapy is no longer effective. One example of this is the loss of estrogen receptor (ER) and progesterone receptor (PR) upon anti-estrogen treatment of breast cancer. [29] Tumors with loss of ER and PR no longer respond to tamoxifen or other anti-estrogen treatments, and while cancer cells remain somewhat responsive to estrogen synthesis inhibitors, they eventually become unresponsive to endocrine manipulation and no longer dependent on estrogen for growth. [29]
Another line of therapeutics used for treating breast cancer is targeting of kinases like human epidermal growth factor receptor 2 (HER2) from the EGFR family. Mutations often occur in the HER2 gene upon treatment with an inhibitor, with about 50% of patients with lung cancer found to have an EGFR-T790M gatekeeper mutation. [12]
Treatment of chronic myeloid leukemia (CML) involves a tyrosine kinase inhibitor that targets the BCR/ABL fusion gene called imatinib. In some people resistant to Imatinib, the BCR/ABL gene is reactivated or amplified, or a single point mutation has occurred on the gene. These point mutations enhance autophosphorylation of the BCR-ABL protein, resulting in the stabilization of the ATP-binding site into its active form, which cannot be bound by imatinib for proper drug activation. [30]
Topoisomerase is a lucrative target for cancer therapy due to its critical role as an enzyme in DNA replication, and many topoisomerase inhibitors have been made. [31] Resistance can occur when topoisomerase levels are decreased, or when different isoforms of topoisomerase are differentially distributed within the cell. Mutant enzymes have also been reported in patient leukemic cells, as well as mutations in other cancers that confer resistance to topoisomerase inhibitors. [31]
Altered metabolism
One of the mechanisms of antineoplastic resistance is over-expression of drug-metabolizing enzymes or carrier molecules. [2] By increasing expression of metabolic enzymes, drugs are more rapidly converted to drug conjugates or inactive forms that can then be excreted. For example, increased expression of glutathione promotes drug resistance, as the electrophilic properties of glutathione allow it to react with cytotoxic agents, inactivating them. [32] In some cases, decreased expression or loss of expression of drug-metabolising enzymes confers resistance, as the enzymes are needed to process a drug from an inactive form to an active form. Arabinoside, a commonly used chemotherapy for leukemia and lymphomas, is converted into cytosine arabinoside triphosphate by deoxycytidine kinase. Mutation of deoxycytidine kinase or loss of expression results in resistance to arabinoside. [2] This is a form of enzymatic deactivation.[ citation needed ]
Growth factor expression levels can also promote resistance to antineoplastic therapies. [2] In breast cancer, drug resistant cells were found to express high levels of IL-6, while sensitive cells did not express significant levels of the growth factor. IL-6 activates the CCAAT enhancer-binding protein transcription factors which activate MDR1 gene expression (see Alteration of Membrane Transport). [33]