
Addison's disease, also known as primary adrenal insufficiency, is a rare long-term endocrine disorder characterized by inadequate production of the steroid hormones cortisol and aldosterone by the two outer layers of the cells of the adrenal glands, causing adrenal insufficiency. Symptoms generally come on slowly and insidiously and may include abdominal pain and gastrointestinal abnormalities, weakness, and weight loss. Darkening of the skin in certain areas may also occur. Under certain circumstances, an adrenal crisis may occur with low blood pressure, vomiting, lower back pain, and loss of consciousness. Mood changes may also occur. Rapid onset of symptoms indicates acute adrenal failure, which is a clinical emergency. An adrenal crisis can be triggered by stress, such as from an injury, surgery, or infection.
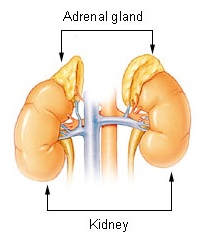
Adrenal insufficiency is a condition in which the adrenal glands do not produce adequate amounts of steroid hormones. The adrenal glands—also referred to as the adrenal cortex—normally secrete glucocorticoids, mineralocorticoids, and androgens. These hormones are important in regulating blood pressure, electrolytes, and metabolism as a whole. Deficiency of these hormones leads to symptoms ranging from abdominal pain, vomiting, muscle weakness and fatigue, low blood pressure, depression, mood and personality changes to organ failure and shock. Adrenal crisis may occur if a person having adrenal insufficiency experiences stresses, such as an accident, injury, surgery, or severe infection; this is a life-threatening medical condition resulting from severe deficiency of cortisol in the body. Death may quickly follow.
Immunodeficiency, also known as immunocompromisation, is a state in which the immune system's ability to fight infectious diseases and cancer is compromised or entirely absent. Most cases are acquired ("secondary") due to extrinsic factors that affect the patient's immune system. Examples of these extrinsic factors include HIV infection and environmental factors, such as nutrition. Immunocompromisation may also be due to genetic diseases/flaws such as SCID.

FOXP3, also known as scurfin, is a protein involved in immune system responses. A member of the FOX protein family, FOXP3 appears to function as a master regulator of the regulatory pathway in the development and function of regulatory T cells. Regulatory T cells generally turn the immune response down. In cancer, an excess of regulatory T cell activity can prevent the immune system from destroying cancer cells. In autoimmune disease, a deficiency of regulatory T cell activity can allow other autoimmune cells to attack the body's own tissues.
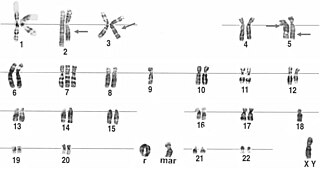
A ring chromosome is an aberrant chromosome whose ends have fused together to form a ring. Ring chromosomes were first discovered by Lilian Vaughan Morgan in 1926. A ring chromosome is denoted by the symbol r in human genetics and R in Drosophila genetics. Ring chromosomes may form in cells following genetic damage by mutagens like radiation, but they may also arise spontaneously during development.
X-linked adrenal hypoplasia congenita is a genetic disorder that mainly affects males. It involves many endocrine tissues in the body, especially the adrenal glands.

Walker–Warburg syndrome (WWS), also called Warburg syndrome, Chemke syndrome, HARD syndrome, Pagon syndrome, cerebroocular dysgenesis (COD) or cerebroocular dysplasia-muscular dystrophy syndrome (COD-MD), is a rare form of autosomal recessive congenital muscular dystrophy. It is associated with brain and eye abnormalities. This condition has a worldwide distribution. Walker-Warburg syndrome is estimated to affect 1 in 60,500 newborns worldwide.
Enteropathy refers to any pathology of the intestine. Although enteritis specifically refers to an inflammation of the intestine, and is thus a more specific term than "enteropathy", the two phrases are sometimes used interchangeably.

Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome is a rare autoimmune disease. It is one of the autoimmune polyendocrine syndromes. Most often, IPEX presents with autoimmune enteropathy, dermatitis (eczema), and autoimmune endocrinopathy, but other presentations exist.

Triple-A syndrome or AAA syndrome is a rare autosomal recessive congenital disorder. In most cases, there is no family history of AAA syndrome. The syndrome was first identified by Jeremy Allgrove and colleagues in 1978; since then just over 100 cases have been reported. The syndrome is called Triple-A due to the manifestation of the illness which includes achalasia, addisonianism, and alacrima. Alacrima is usually the earliest manifestation. Neurodegeneration or atrophy of the nerve cells and autonomic dysfunction may be seen in the disorder; therefore, some have suggested the disorder be called 4A syndrome. It is a progressive disorder that can take years to develop the full-blown clinical picture. The disorder also has variability and heterogeneity in presentation.
Acrodermatitis enteropathica is an autosomal recessive metabolic disorder affecting the uptake of zinc through the inner lining of the bowel, the mucous membrane. It is characterized by inflammation of the skin (dermatitis) around bodily openings (periorificial) and the tips of fingers and toes (acral), hair loss (alopecia), and diarrhea. It can also be related to deficiency of zinc due to other, i.e. congenital causes.

Bare lymphocyte syndrome type II is a rare recessive genetic condition in which a group of genes called major histocompatibility complex class II are not expressed. The result is that the immune system is severely compromised and cannot effectively fight infection.
Immune dysregulation is any proposed or confirmed breakdown or maladaptive change in molecular control of immune system processes. For example, dysregulation is a component in the pathogenesis of autoimmune diseases and some cancers. Immune system dysfunction, as seen in IPEX syndrome leads to immune dysfunction, polyendocrinopathy, enteropathy, X-linked (IPEX). IPEX typically presents during the first few months of life with diabetes mellitus, intractable diarrhea, failure to thrive, eczema, and hemolytic anemia. unrestrained or unregulated immune response.

The autoimmune regulator (AIRE) is a protein that in humans is encoded by the AIRE gene. It is a 13kb gene on chromosome 21q22.3 that has 545 amino acids. AIRE is a transcription factor expressed in the medulla of the thymus. It is part of the mechanism which eliminates self-reactive T cells that would cause autoimmune disease. It exposes T cells to normal, healthy proteins from all parts of the body, and T cells that react to those proteins are destroyed.
Allan–Herndon–Dudley syndrome is a rare X-linked inherited disorder of brain development that causes both moderate to severe intellectual disability and problems with speech and movement.

Chronic mucocutaneous candidiasis is an immune disorder of T cells. It is characterized by chronic infections with Candida that are limited to mucosal surfaces, skin, and nails. It can also be associated with other types of infections, such as human papilloma virus. An association with chromosome 2 has been identified.
Autoimmune polyendocrine syndrome type 2, a form of autoimmune polyendocrine syndrome also known as APS-II, or PAS II, is the most common form of the polyglandular failure syndromes. PAS II is defined as the association between autoimmune Addison's disease and either autoimmune thyroid disease, type 1 diabetes, or both. It is heterogeneous and has not been linked to one gene. Rather, individuals are at a higher risk when they carry a particular human leukocyte antigen. APS-II affects women to a greater degree than men.
Autoimmune polyendocrine syndrome type 1 (APS-1), is a subtype of autoimmune polyendocrine syndrome. It causes the dysfunction of multiple endocrine glands due to autoimmunity. It is a genetic disorder, inherited in autosomal recessive fashion due to a defect in the AIRE gene , which is located on chromosome 21 and normally confers immune tolerance.

Autoimmune enteropathy is a rare autoimmune disorder characterized by weight loss from malabsorption, severe and protracted diarrhea, and autoimmune damage to the intestinal mucosa. Autoimmune enteropathy typically occurs in infants and younger children however, adult cases have been reported in literature. Autoimmune enteropathy was first described by Walker-Smith et al. in 1982.
Autoimmune polyendocrine syndrome, type 3 is characterized by the coexistence of two autoimmune illnesses, not including Addison's disease, and an autoimmune thyroid disease. Based on other organ-specific autoimmune involvement, there are multiple subtypes that are classified: type 3a shows thyroid autoimmune disease in conjunction with type 1 diabetes, type 3b shows thyroid autoimmune disease in conjunction with pernicious anemia (PA), and type 3c shows thyroid autoimmune disease in conjunction with alopecia, vitiligo, or other organ-specific autoimmune disease.


