| Protein | Pathway | Description |
|---|
| ATR | Nucleotide excision repair [1] | deletion of ATR in adult mice leads to a number of disorders including hair loss and graying, kyphosis, osteoporosis, premature involution of the thymus, fibrosis of the heart and kidney and decreased spermatogenesis [2] |
| DNA-PKcs | Non-homologous end joining | shorter lifespan, earlier onset of aging related pathologies; [3] [4] higher level of DNA damage persistence [5] |
| ERCC1 | Nucleotide excision repair, Interstrand cross link repair [6] | deficient transcription coupled NER with time-dependent accumulation of transcription-blocking damages; [7] mouse life span reduced from 2.5 years to 5 months; [8] ) Ercc1−/− mice are leukopenic and thrombocytopenic, and there is extensive adipose transformation of the bone marrow, hallmark features of normal aging in mice [6] |
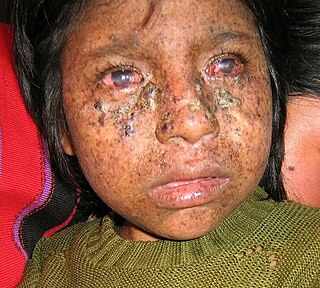
| ERCC2 (XPD) | Nucleotide excision repair (also transcription as part of TFIIH) | some mutations in ERCC2 cause Cockayne syndrome in which patients have segmental progeria with reduced stature, intellectual disability, cachexia (loss of subcutaneous fat tissue), sensorineural deafness, retinal degeneration, and calcification of the central nervous system; other mutations in ERCC2 cause trichothiodystrophy in which patients have segmental progeria with brittle hair, short stature, progressive cognitive impairment and abnormal face shape; still other mutations in ERCC2 cause xeroderma pigmentosum (without a progeroid syndrome) and with extreme sun-mediated skin cancer predisposition [9] |
| ERCC4 (XPF) | Nucleotide excision repair, Interstrand cross link repair, Single-strand annealing, Microhomology-mediated end joining [6] | mutations in ERCC4 cause symptoms of accelerated aging that affect the neurologic, hepatobiliary, musculoskeletal, and hematopoietic systems, and cause an old, wizened appearance, loss of subcutaneous fat, liver dysfunction, vision and hearing loss, chronic kidney disease, muscle wasting, osteopenia, kyphosis and cerebral atrophy [6] |
| ERCC5 (XPG) | Nucleotide excision repair, [10] Homologous recombinational repair, [11] Base excision repair [12] [13] | mice with deficient ERCC5 show loss of subcutaneous fat, kyphosis, osteoporosis, retinal photoreceptor loss, liver aging, extensive neurodegeneration, and a short lifespan of 4–5 months |
| ERCC6 (Cockayne syndrome B or CS-B) | Nucleotide excision repair [especially transcription coupled repair (TC-NER) and interstrand crosslink repair] | premature aging features with shorter life span and photosensitivity, [14] deficient transcription coupled NER with accumulation of unrepaired DNA damages, [15] also defective repair of oxidatively generated DNA damages including 8-oxoguanine, 5-hydroxycytosine and cyclopurines [15] |
| ERCC8 (Cockayne syndrome A or CS-A) | Nucleotide excision repair [especially transcription coupled repair (TC-NER) and interstrand crosslink repair] | premature aging features with shorter life span and photosensitivity, [14] deficient transcription coupled NER with accumulation of unrepaired DNA damages, [15] also defective repair of oxidatively generated DNA damages including 8-oxoguanine, 5-hydroxycytosine and cyclopurines [15] |
| GTF2H5 (TTDA) | Nucleotide excision repair | deficiency causes trichothiodystrophy (TTD) a premature-ageing and neuroectodermal disease; humans with GTF2H5 mutations have a partially inactivated protein [16] with retarded repair of 6-4-photoproducts [17] |
| Ku70 | Non-homologous end joining | shorter lifespan, earlier onset of aging related pathologies; [18] persistent foci of DNA double-strand break repair proteins [19] |
| Ku80 | Non-homologous end joining | shorter lifespan, earlier onset of aging related pathologies; [20] defective repair of spontaneous DNA damage [18] |
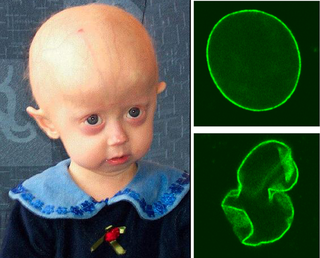
| Lamin A | Non-homologous end joining, Homologous recombination | increased DNA damage and chromosome aberrations; progeria; aspects of premature aging; altered expression of numerous DNA repair factors [21] |
| NRMT1 | Nucleotide excision repair [22] | mutation in NRMT1 causes decreased body size, female-specific infertility, kyphosis, decreased mitochondrial function, and early-onset liver degeneration [23] |
| RECQL4 | Base excision repair, Nucleotide excision repair, Homologous recombination, Non-homologous end joining [24] | mutations in RECQL4 cause Rothmund-Thomson syndrome, with alopecia, sparse eyebrows and lashes, cataracts and osteoporosis [24] |
| SIRT6 | Base excision repair, Nucleotide excision repair, Homologous recombination, Non-homologous end joining [25] | SIRT6-deficient mice develop profound lymphopenia, loss of subcutaneous fat and lordokyphosis, and these defects overlap with aging-associated degenerative processes [26] |
| SIRT7 | Non-homologous end joining | mice defective in SIRT7 show phenotypic and molecular signs of accelerated aging such as premature pronounced curvature of the spine, reduced life span, and reduced non-homologous end joining [27] |
| Werner syndrome helicase | Homologous recombination, [28] [29] Non-homologous end joining, [30] Base excision repair, [31] [32] Replication arrest recovery [33] | shorter lifespan, earlier onset of aging related pathologies, genome instability [34] [35] |
| ZMPSTE24 | Homologous recombination | lack of Zmpste24 prevents lamin A formation and causes progeroid phenotypes in mice and humans, increased DNA damage and chromosome aberrations, sensitivity to DNA-damaging agents and deficiency in homologous recombination [36] |