




DNA repair protein complementing XP-G cells is a protein that in humans is encoded by the ERCC5 gene. [5] [6]
DNA repair protein complementing XP-G cells is a protein that in humans is encoded by the ERCC5 gene. [5] [6]
Excision repair cross-complementing rodent repair deficiency, complementation group 5 (xeroderma pigmentosum, complementation group G) is involved in excision repair of UV-induced DNA damage. Mutations cause Cockayne syndrome, which is characterized by severe growth defects, mental retardation, and cachexia. Multiple alternatively spliced transcript variants encoding distinct isoforms have been described, but the biological validity of all variants has not been determined. [6]
Mutations in ERCC5 cause arthrogryposis. [7]
XPG is a structure specific endonuclease that incises DNA at the 3’ side of the damaged nucleotide during nucleotide excision repair.
Mutational defects in the Ercc5(Xpg) gene can cause either the cancer-prone condition xeroderma pigmentosum (XP) alone, or in combination with the severe neurodevelopmental disorder Cockayne syndrome (CS) or the infantile lethal cerebro-oculo-facio-skeletal syndrome. [8]
An Ercc5(Xpg) mutant mouse model presented features of premature aging including cachexia and osteoporosis with pronounced degenerative phenotypes in both liver and brain. [8] These mutant mice developed a multi-system premature aging degenerative phenotype that appears to strengthen the link between DNA damage and aging. [8] (see DNA damage theory of aging).
Dietary restriction, which extends lifespan of wild-type mice, also substantially increased the lifespan of Ercc5(Xpg) mutant mice. [9] Dietary restriction of the mutant mice, while delaying aging, also appeared to slow the accumulation of genome wide DNA damage and to preserve transcriptional output, thus contributing to improved cell viability.
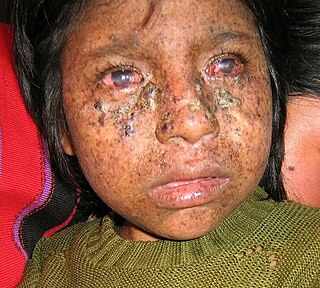
Xeroderma pigmentosum (XP) is a genetic disorder in which there is a decreased ability to repair DNA damage such as that caused by ultraviolet (UV) light. Symptoms may include a severe sunburn after only a few minutes in the sun, freckling in sun-exposed areas, dry skin and changes in skin pigmentation. Nervous system problems, such as hearing loss, poor coordination, loss of intellectual function and seizures, may also occur. Complications include a high risk of skin cancer, with about half having skin cancer by age 10 without preventative efforts, and cataracts. There may be a higher risk of other cancers such as brain cancers.

Nucleotide excision repair is a DNA repair mechanism. DNA damage occurs constantly because of chemicals, radiation and other mutagens. Three excision repair pathways exist to repair single stranded DNA damage: Nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR). While the BER pathway can recognize specific non-bulky lesions in DNA, it can correct only damaged bases that are removed by specific glycosylases. Similarly, the MMR pathway only targets mismatched Watson-Crick base pairs.

XPB is an ATP-dependent DNA helicase in humans that is a part of the TFIIH transcription factor complex.
A DNA repair-deficiency disorder is a medical condition due to reduced functionality of DNA repair.
Richard D. Wood is an American molecular biologist specializing in research on DNA repair and mutation. He is known for pioneering studies on nucleotide excision repair (NER), particularly for reconstituting the minimum set of proteins involved in this process, identifying proliferating cell nuclear antigen (PCNA) as part of the NER complex and identifying mammalian repair polymerases.

ERCC2, or XPD is a protein involved in transcription-coupled nucleotide excision repair.
Transcription factor II H (TFIIH) is an important protein complex, having roles in transcription of various protein-coding genes and DNA nucleotide excision repair (NER) pathways. TFIIH first came to light in 1989 when general transcription factor-δ or basic transcription factor 2 was characterized as an indispensable transcription factor in vitro. This factor was also isolated from yeast and finally named TFIIH in 1992.
Excision repair cross-complementing (ERCC) is a set of proteins which are involved in DNA repair.

DNA excision repair protein ERCC-1 is a protein that in humans is encoded by the ERCC1 gene. Together with ERCC4, ERCC1 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.

DNA damage-binding protein 2 is a protein that in humans is encoded by the DDB2 gene.

UV excision repair protein RAD23 homolog B is a protein that in humans is encoded by the RAD23B gene.
The enzyme DNA-(apurinic or apyrimidinic site) lyase, also referred to as DNA-(apurinic or apyrimidinic site) 5'-phosphomonoester-lyase or DNA AP lyase catalyzes the cleavage of the C-O-P bond 3' from the apurinic or apyrimidinic site in DNA via β-elimination reaction, leaving a 3'-terminal unsaturated sugar and a product with a terminal 5'-phosphate. In the 1970s, this class of enzyme was found to repair at apurinic or apyrimidinic DNA sites in E. coli and in mammalian cells. The major active enzyme of this class in bacteria, and specifically, E. coli is endonuclease type III. This enzyme is part of a family of lyases that cleave carbon-oxygen bonds.

Xeroderma pigmentosum, complementation group C, also known as XPC, is a protein which in humans is encoded by the XPC gene. XPC is involved in the recognition of bulky DNA adducts in nucleotide excision repair. It is located on chromosome 3.

DNA repair protein complementing XP-A cells is a protein that in humans is encoded by the XPA gene.

DNA excision repair protein ERCC-6 is a protein that in humans is encoded by the ERCC6 gene. The ERCC6 gene is located on the long arm of chromosome 10 at position 11.23.
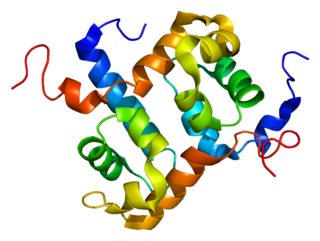
ERCC4 is a protein designated as DNA repair endonuclease XPF that in humans is encoded by the ERCC4 gene. Together with ERCC1, ERCC4 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.

DNA excision repair protein ERCC-8 is a protein that in humans is encoded by the ERCC8 gene.
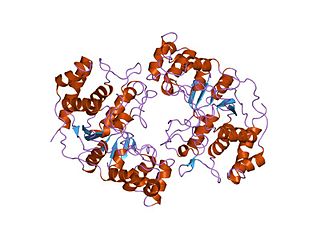
In molecular biology the protein domain XPG refers to, in this case, the N-terminus of XPG. The XPG protein can be corrected by a 133 kDa nuclear protein, XPGC. XPGC is an acidic protein that confers normal ultraviolet (UV) light resistance. It is a magnesium-dependent, single-strand DNA endonuclease that makes structure-specific endonucleolytic incisions in a DNA substrate containing a duplex region and single-stranded arms. XPGC cleaves one strand of the duplex at the border with the single-stranded region.

In molecular biology, the XPG-I is a protein domain found on Xeroderma Pigmentosum Complementation Group G (XPG) protein. The XPG protein is an endonuclease which repairs DNA damage caused by ultraviolet light. The XPG protein repairs DNA by a process called, Nucleotide excision repair. Mutations in the protein commonly cause Xeroderma Pigmentosum which often lead to skin cancer.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.
| Excision repair | |
|---|---|
| Other forms of repair | |
| Other/ungrouped proteins | |
| Regulation | |
| Other/ungrouped | |