Fanconi anaemia (FA) is a rare genetic disease resulting in impaired response to DNA damage. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), and 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity.

Helicases are a class of enzymes thought to be vital to all organisms. Their main function is to unpack an organism's genetic material. Helicases are motor proteins that move directionally along a nucleic acid phosphodiester backbone, separating two hybridized nucleic acid strands, using energy from ATP hydrolysis. There are many helicases, representing the great variety of processes in which strand separation must be catalyzed. Approximately 1% of eukaryotic genes code for helicases.
RecQ helicase is a family of helicase enzymes initially found in Escherichia coli that has been shown to be important in genome maintenance. They function through catalyzing the reaction ATP + H2O → ADP + P and thus driving the unwinding of paired DNA and translocating in the 3' to 5' direction. These enzymes can also drive the reaction NTP + H2O → NDP + P to drive the unwinding of either DNA or RNA.

Bloom syndrome is a rare autosomal recessive genetic disorder characterized by short stature, predisposition to the development of cancer, and genomic instability. BS is caused by mutations in the BLM gene which is a member of the RecQ DNA helicase family. Mutations in other members of this family, namely WRN and RECQL4, are associated with the clinical entities Werner syndrome and Rothmund–Thomson syndrome, respectively. More broadly, Bloom syndrome is a member of a class of clinical entities that are characterized by chromosomal instability, genomic instability, or both and by cancer predisposition.
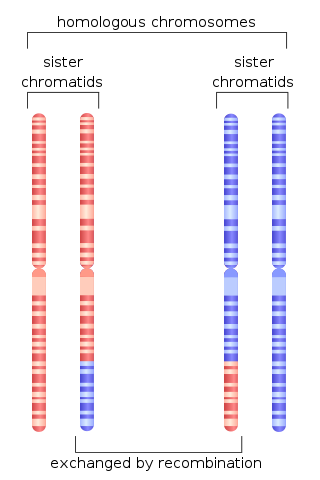
Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids.

Bloom syndrome protein is a protein that in humans is encoded by the BLM gene and is not expressed in Bloom syndrome.

Fanconi anaemia, complementation group A, also known as FAA, FACA and FANCA, is a protein which in humans is encoded by the FANCA gene. It belongs to the Fanconi anaemia complementation group (FANC) family of genes of which 12 complementation groups are currently recognized and is hypothesised to operate as a post-replication repair or a cell cycle checkpoint. FANCA proteins are involved in inter-strand DNA cross-link repair and in the maintenance of normal chromosome stability that regulates the differentiation of haematopoietic stem cells into mature blood cells.

Fanconi anemia group D2 protein is a protein that in humans is encoded by the FANCD2 gene. The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN and FANCO.

Fanconi anemia group G protein is a protein that in humans is encoded by the FANCG gene.

Exonuclease 1 is an enzyme that in humans is encoded by the EXO1 gene.
DNA repair and recombination protein RAD54-like is a protein that in humans is encoded by the RAD54L gene.

Fanconi anemia group J protein is a protein that in humans is encoded by the BRCA1-interacting protein 1 (BRIP1) gene.

Fanconi anemia group F protein is a protein that in humans is encoded by the FANCF gene.
DNA topoisomerase 3-alpha is an enzyme that in humans is encoded by the TOP3A gene.

E3 ubiquitin-protein ligase FANCL is an enzyme that in humans is encoded by the FANCL gene.
Structural maintenance of chromosomes protein 6 is a protein that in humans is encoded by the SMC6 gene.

Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene.

RecQ-mediated genome instability protein 1 is a protein that in humans is encoded by the RMI1 gene.
Sgs1, also known as slow growth suppressor 1, is a DNA helicase protein found in Saccharomyces cerevisiae. It is a homolog of the bacterial RecQ helicase. Like the other members of the RecQ helicase family, Sgs1 is important for DNA repair. In particular, Sgs1 collaborates with other proteins to repair double-strand breaks during homologous recombination in eukaryotes.

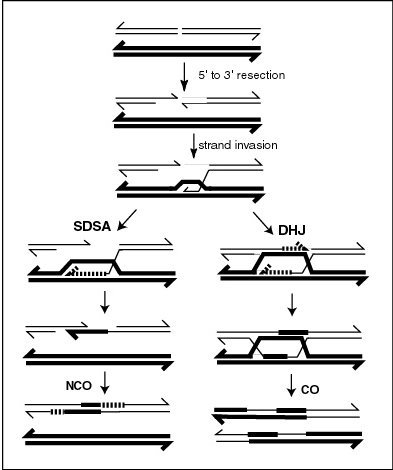
Synthesis-dependent strand annealing (SDSA) is a major mechanism of homology-directed repair of DNA double-strand breaks (DSBs). Although many of the features of SDSA were first suggested in 1976, the double-Holliday junction model proposed in 1983 was favored by many researchers. In 1994, studies of double-strand gap repair in Drosophila were found to be incompatible with the double-Holliday junction model, leading researchers to propose a model they called synthesis-dependent strand annealing. Subsequent studies of meiotic recombination in S. cerevisiae found that non-crossover products appear earlier than double-Holliday junctions or crossover products, challenging the previous notion that both crossover and non-crossover products are produced by double-Holliday junctions and leading the authors to propose that non-crossover products are generated through SDSA.