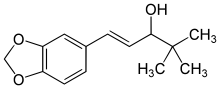
Valproate (VPA) and its valproic acid, sodium valproate, and valproate semisodium forms are medications primarily used to treat epilepsy and bipolar disorder and prevent migraine headaches. They are useful for the prevention of seizures in those with absence seizures, partial seizures, and generalized seizures. They can be given intravenously or by mouth, and the tablet forms exist in both long- and short-acting formulations.
Anticonvulsants are a diverse group of pharmacological agents used in the treatment of epileptic seizures. Anticonvulsants are also increasingly being used in the treatment of bipolar disorder and borderline personality disorder, since many seem to act as mood stabilizers, and for the treatment of neuropathic pain. Anticonvulsants suppress the excessive rapid firing of neurons during seizures. Anticonvulsants also prevent the spread of the seizure within the brain.

Lamotrigine, sold under the brand name Lamictal among others, is a medication used to treat epilepsy and stabilize mood in bipolar disorder. For epilepsy, this includes focal seizures, tonic-clonic seizures, and seizures in Lennox-Gastaut syndrome. In bipolar disorder, lamotrigine has not been shown to reliably treat acute depression; but for patients with bipolar disorder who are not currently symptomatic, it appears to be effective in reducing the risk of future episodes of depression.
Absence seizures are one of several kinds of generalized seizures. These seizures are sometimes referred to as petit mal seizures. Absence seizures are characterized by a brief loss and return of consciousness, generally not followed by a period of lethargy. Absence seizure is very common in children. It affects both sides of the brain.

Myoclonus is a brief, involuntary, irregular twitching of a muscle or a group of muscles, different from clonus, which is rhythmic or regular. Myoclonus describes a medical sign and, generally, is not a diagnosis of a disease. These myoclonic twitches, jerks, or seizures are usually caused by sudden muscle contractions or brief lapses of contraction. The most common circumstance under which they occur is while falling asleep. Myoclonic jerks occur in healthy people and are experienced occasionally by everyone. However, when they appear with more persistence and become more widespread they can be a sign of various neurological disorders. Hiccups are a kind of myoclonic jerk specifically affecting the diaphragm. When a spasm is caused by another person it is known as a provoked spasm. Shuddering attacks in babies fall in this category.

Clonazepam, sold under the brand name Klonopin among others, is a medication used to prevent and treat seizures, panic disorder, anxiety, and the movement disorder known as akathisia. It is a tranquilizer of the benzodiazepine class. It possesses anxiolytic, anticonvulsant, sedative, hypnotic, and skeletal muscle relaxant properties. It is typically taken by mouth. Effects begin within one hour and last between six and twelve hours.

Tiagabine is an anticonvulsant medication produced by Cephalon that is used in the treatment of epilepsy. The drug is also used off-label in the treatment of anxiety disorders and panic disorder.

Primidone, sold under various brand names, is a barbiturate medication that is used to treat partial and generalized seizures, as well as essential tremors. It is taken by mouth.

Lennox–Gastaut syndrome (LGS) is a complex, rare, and severe childhood-onset epilepsy. It is characterized by multiple and concurrent seizure types, cognitive dysfunction, and slow spike waves on electroencephalogram (EEG). Typically, it presents in children aged 3–5 years and can persist into adulthood. It has been associated with several gene mutations, perinatal injuries, congenital infections, brain tumors/malformations, and genetic disorders such as tuberous sclerosis and West syndrome. The prognosis for LGS is poor with a 5% mortality in childhood and persistent seizures into adulthood (80%–90%).

Clobazam, sold under the brand names Frisium, Onfi and others, is a benzodiazepine class medication that was patented in 1968. Clobazam was first synthesized in 1966 and first published in 1969. Clobazam was originally marketed as an anxioselective anxiolytic since 1970, and an anticonvulsant since 1984. The primary drug-development goal was to provide greater anxiolytic, anti-obsessive efficacy with fewer benzodiazepine-related side effects.
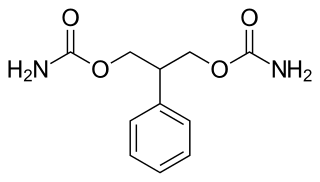
Felbamate is an anticonvulsant used in the treatment of epilepsy. It is used to treat partial seizures in adults and partial and generalized seizures associated with Lennox–Gastaut syndrome in children. However, an increased risk of potentially fatal aplastic anemia and/or liver failure limit the drug's usage to severe refractory epilepsy.

Sultiame is a sulfonamide and inhibitor of the enzyme carbonic anhydrase. It is used as an anticonvulsant.
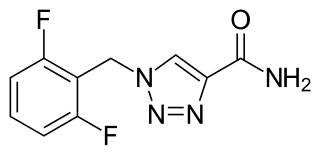
Rufinamide is an anticonvulsant medication. It is used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders. Rufinamide, a triazole derivative, was developed in 2004 by Novartis Pharma, AG, and is manufactured by Eisai.
Generalized epilepsy with febrile seizures plus (GEFS+) is a syndromic autosomal dominant disorder where affected individuals can exhibit numerous epilepsy phenotypes. GEFS+ can persist beyond early childhood. GEFS+ is also now believed to encompass three other epilepsy disorders: severe myoclonic epilepsy of infancy (SMEI), which is also known as Dravet's syndrome, borderline SMEI (SMEB), and intractable epilepsy of childhood (IEC). There are at least six types of GEFS+, delineated by their causative gene. Known causative gene mutations are in the sodium channel α subunit genes SCN1A, an associated β subunit SCN1B, and in a GABAA receptor γ subunit gene, in GABRG2 and there is another gene related with calcium channel the PCDH19 which is also known as Epilepsy Female with Mental Retardation. Penetrance for this disorder is estimated at 60%.
Dravet syndrome, previously known as severe myoclonic epilepsy of infancy (SMEI), is an autosomal dominant genetic disorder which causes a catastrophic form of epilepsy, with prolonged seizures that are often triggered by hot temperatures or fever. It is very difficult to treat with anticonvulsant medications. It often begins before 1 year of age, with 6 months being the age that seizures, characterized by prolonged convulsions and triggered by fever, usually begin.
Idiopathic generalized epilepsy (IGE) is a group of epileptic disorders that are believed to have a strong underlying genetic basis. Patients with an IGE subtype are typically otherwise normal and have no structural brain abnormalities. People also often have a family history of epilepsy and seem to have a genetically predisposed risk of seizures. IGE tends to manifest itself between early childhood and adolescence although it can be eventually diagnosed later. The genetic cause of some IGE types is known, though inheritance does not always follow a simple monogenic mechanism.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).

Etifoxine is an anxiolytic and anticonvulsant drug developed by Hoechst in the 1960s. It is sold in approximately 40 countries for anxiety disorders, without the sedation and ataxia associated with benzodiazepine drugs. It has similar anxiolytic effects to benzodiazepine drugs, but is structurally distinct, although it has structural elements in common with them. Studies suggest is as effective as lorazepam as an anxiolytic, but has fewer side effects. Etifoxine is not approved by the U.S. Food and Drug Administration. The European Medicines Agency (EMA) started a review procedure regarding the effectiveness and safety of etifoxine following a French study that compares etifoxine's effectiveness to placebo and lorazepam. In January 2022, the EMA "finalized its review of Stresam and concluded that the medicine can continue to be used for the treatment of anxiety disorders, but it must not be used in patients who previously had severe skin reactions or severe liver problems after taking etifoxine."
Early myoclonic encephalopathy (EME) is a rare neonatal-onset epilepsy developmental and epileptic encephalopathy (DEE) with an onset at neonatal period or during the first 3 months of life. It is marked by the presence of myoclonic seizures but multiple seizure types may occur. The electroencephalographic recording is abnormal with eitherusually a suppression-burst pattern or other significantly abnormal patterns. On most occasions the seizures are drug-resistant. After several months, the seizure pattern may develop into infantile spasms syndrome. The neurological exam is abnormal with a significant risk of early death. Various genetic and metabolic disorders are responsible. At present, EME and Ohtahara syndrome are recorded as distinct patterns in the categorization of epilepsies but both neonatal-onset epilepsy syndromes are considered to be merged in one unique entity. It is a severe type of epilepsy syndrome associated with high level of resistance to treatment and a high risk for cognitive impairment. The myoclonic seizures could be seen in other epilepsy syndromes. Multiple types of childhood epilepsies are usually mentioned as myoclonic epilepsies when the myoclonic seizures are a predominant feature.
People with epilepsy may be classified into different syndromes based on specific clinical features. These features include the age at which seizures begin, the seizure types, and EEG findings, among others. Identifying an epilepsy syndrome is useful as it helps determine the underlying causes as well as deciding what anti-seizure medication should be tried. Epilepsy syndromes are more commonly diagnosed in infants and children. Some examples of epilepsy syndromes include benign rolandic epilepsy, childhood absence epilepsy and juvenile myoclonic epilepsy. Severe syndromes with diffuse brain dysfunction caused, at least partly, by some aspect of epilepsy, are also referred to as epileptic encephalopathies. These are associated with frequent seizures that are resistant to treatment and severe cognitive dysfunction, for instance Lennox-Gastaut syndrome and West syndrome.