1,4-alpha-glucan-branching enzyme, also known as brancher enzyme or glycogen-branching enzyme is an enzyme that in humans is encoded by the GBE1gene.[5]
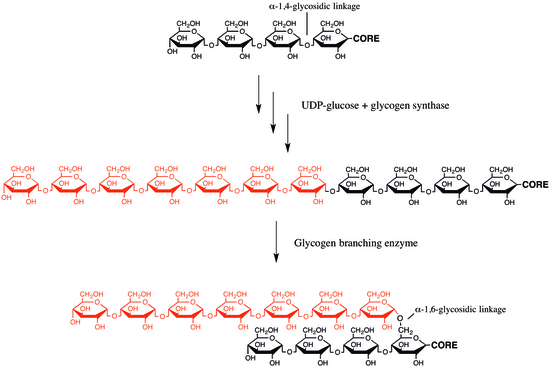
Glycogen branching enzyme is an enzyme that adds branches to the growing glycogen molecule during the synthesis of glycogen, a storage form of glucose. More specifically, during glycogen synthesis, a glucose 1-phosphate molecule reacts with uridine triphosphate (UTP) to become UDP-glucose, an activated form of glucose. The activated glucosyl unit of UDP-glucose is then transferred to the hydroxyl group at the C-4 of a terminal residue of glycogen to form an α-1,4-glycosidic linkage, a reaction catalyzed by glycogen synthase. Importantly, glycogen synthase can only catalyze the synthesis of α-1,4-glycosidic linkages. Since glycogen is a readily mobilized storage form of glucose, the extended glycogen polymer is branched by glycogen branching enzyme to provide glycogen breakdown enzymes, such as glycogen phosphorylase, with many terminal residues for rapid degradation. Branching also importantly increases the solubility and decreases the osmotic strength of glycogen.[6]
The protein encoded by this gene is a glycogen branching enzyme that catalyzes the transfer of alpha-1,4-linked glucosyl units from the outer end of a glycogen chain to an alpha-1,6 position on the same or a neighboring glycogen chain. Branching of the chains is essential to increase the solubility of the glycogen molecule and, consequently, in reducing the osmotic pressure within cells. The highest levels of this enzyme are found in liver and muscle cells. Mutations in this gene are associated with glycogen storage disease type IV (also known as Andersen's disease).
Nomenclature
This enzyme belongs to the family of transferases, to be specific, those glycosyltransferases that transfer hexoses (hexosyltransferases). The systematic name of this enzyme class is 1,4-alpha-D-glucan:1,4-alpha-D-glucan 6-alpha-D-(1,4-alpha-D-glucano)-transferase. Other names in common use include branching enzyme, amylo-(1,4→1,6)-transglycosylase, Q-enzyme, alpha-glucan-branching glycosyltransferase, amylose isomerase, enzymatic branching factor, branching glycosyltransferase, enzyme Q, glucosan transglycosylase, 1,4-alpha-glucan branching enzyme 1, plant branching enzyme, alpha-1,4-glucan:alpha-1,4-glucan-6-glycosyltransferase, and starch branching enzyme. This enzyme participates in starch and sucrose metabolism.
Through Southern blot analysis of DNA derived from human/rodent somatic cell hybrids, GBE1 has been identified as an autosomal gene located on the short arm of chromosome 3 at position 12.3.[7][8][9][10] The human GBE gene was also isolated by a function complementation of the Saccharomyces cerevisiae GBE deficiency.[10] From the isolated cDNA, the length of the gene was found to be approximately 3 kb.[10] Additionally, the coding sequence was found to comprise 2,106 base pairs and encode a 702-amino acid long GBE. The molecular mass of human GBE was calculated to be 80,438 Da.[10]
Structure
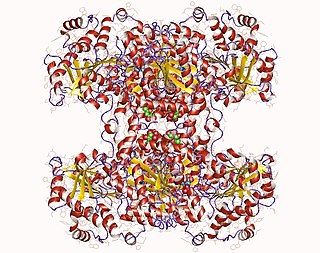
Structure of glycogen branching enzyme found in E. Coli
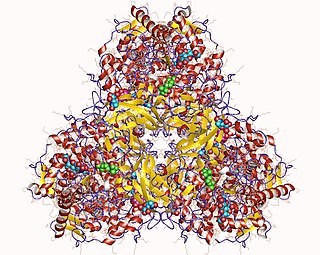
Glycogen branching enzyme belongs to the α-amylase family of enzymes, which include α-amylases, pullulanas/isoamylase, cyclodextrin glucanotransferase (CGT), and branching enzyme.[11][12] Shown by x-ray crystallography, glycogen branching enzyme has four marginally asymmetric units each that are organized into three domains: an amino-terminal domain, involved in determining the length of the chain transfer, a carboxyl-terminal domain, involved in substrate preference and catalytic capacity, and a central (α/β) barrel catalytic domain.[11][13][14][15] The amino-terminal domain consists of 128 residues arranged in seven β-strands, the carboxyl-terminal domain with 116 residues also organized in seven β-strands, and the (α/β) barrel domain with 372 residues. While the central (α/β) barrel domain is common in members of the α-amylase family, numerous variations exist between the various barrel domains. Additionally, there are striking differences between the loops connecting elements of the secondary structure among these various α-amylase members, especially around the active site. In comparison to the other family members, glycogen binding enzyme has shorter loops, which result in a more open cavity, favorable to the binding of a bulkier substrate such as branched sugar. Through primary structure analysis and the x-ray crystallographic structures of the members of the α-amylase family, seven residue were conserved, Asp335, His340, Arg403, Asp 405, Glu458, His525, and Asp526 (E coli. numbering). These residues are implicated in catalysis and substrate binding.[11]
Glycogen binding enzymes in other organisms have also been crystallized and structurally determined, demonstrating both similarity and variation to GBE found in Escherichia coli.[16][17][18][19]
Function
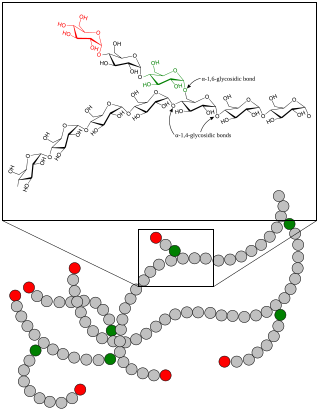
Scheme demonstrating the function of glycogen branching enzyme
In glycogen, every 10 to 14 glucose units, a side branch with an additional chain of glucose units occurs. The side chain attaches at carbon atom 6 of a glucose unit, an α-1,6-glycosidic bond. This connection is catalyzed by a branching enzyme, generally given the name α-glucan branching enzyme. A branching enzyme attaches a string of seven glucose units (with some minor variation to this number) to the carbon at the C-6 position on the glucose unit, forming the α-1,6-glycosidic bond. The specific nature of this enzyme means that this chain of 7 carbons is usually attached to a glucose molecule that is in position three from the non-reducing end of another chain. Because the enzyme works with such specificity regarding the number of glucose units transferred and the position to which they are transferred, the enzyme creates the very characteristic, highly branched glycogen molecule.[20]
Approximately 40 mutations in the GBE1 gene, most resulting in a point mutation in the glycogen branching enzyme, have led to the early childhood disorder, glycogen storage disease type IV (GSD IV).[9] This disease is characterized by a severe depletion or complete absence of GBE, resulting in the accumulation of abnormally structured glycogen, known as polyglucosan bodies.[9] Glycogen buildup leads to increased osmotic pressure resulting in cellular swelling and death.[9] The tissues most affected by this disease are the liver, heart, and neuromuscular system, areas with the greatest levels of glycogen accumulation.[9][22] Abnormal glycogen buildup in the liver interferes with liver functioning and can result in an enlarged liver and liver disease.[9][23] In muscles, the inability of cells to efficiently breakdown glycogen due to the severe reduction or absence of branching can lead to muscle weakness and atrophy.[9] At least three mutations in the GBE1 gene have been found to cause another disease called adult polyglucosan body disease (APBD).[9][24] While in GSD IV GBE activity is undetectable or minimally detectable, APBD is characterized by reduced or even normal GBE activity.[24] In this disease, abnormal glycogen can build up in neurons leading to a spectrum of problems. Specifically, some disease characteristics are gait difficulties from mixed upper and lower motor neuron involvement sensory loss in lower extremities, and neurogenic bladder, a problem in which a person lacks bladder control due to a brain, spinal cord, or nerve condition.[24][25]
Male and female animals underwent a standardized phenotypic screen to determine the effects of deletion.[27][34] Twenty six tests were carried out on mutant mice and two significant abnormalities were observed.[27] No homozygousmutant embryos were identified during gestation, and therefore none survived until weaning. The remaining tests were carried out on heterozygous mutant adult mice; no additional significant abnormalities were observed in these animals.[27]
Related Research Articles
An amylase is an enzyme that catalyses the hydrolysis of starch into sugars. Amylase is present in the saliva of humans and some other mammals, where it begins the chemical process of digestion. Foods that contain large amounts of starch but little sugar, such as rice and potatoes, may acquire a slightly sweet taste as they are chewed because amylase degrades some of their starch into sugar. The pancreas and salivary gland make amylase to hydrolyse dietary starch into disaccharides and trisaccharides which are converted by other enzymes to glucose to supply the body with energy. Plants and some bacteria also produce amylase. Specific amylase proteins are designated by different Greek letters. All amylases are glycoside hydrolases and act on α-1,4-glycosidic bonds.
Maltase is one type of alpha-glucosidase enzymes located in the brush border of the small intestine. This enzyme catalyzes the hydrolysis of disaccharide maltose into two simple sugars of glucose. Maltase is found in plants, bacteria, yeast, humans, and other vertebrates. It is thought to be synthesized by cells of the mucous membrane lining the intestinal wall.
Glycogen phosphorylase is one of the phosphorylase enzymes. Glycogen phosphorylase catalyzes the rate-limiting step in glycogenolysis in animals by releasing glucose-1-phosphate from the terminal alpha-1,4-glycosidic bond. Glycogen phosphorylase is also studied as a model protein regulated by both reversible phosphorylation and allosteric effects.
Glycogen-branching enzyme deficiency (GBED) is an inheritable glycogen storage disease affecting American Quarter Horses and American Paint Horses. It leads to abortion, stillbirths, or early death of affected animals. The human form of the disease is known as glycogen storage disease type IV.
Glycogen synthase is a key enzyme in glycogenesis, the conversion of glucose into glycogen. It is a glycosyltransferase that catalyses the reaction of UDP-glucose and n to yield UDP and n+1.
The glycogen debranching enzyme, in humans, is the protein encoded by the gene AGL. This enzyme is essential for the breakdown of glycogen, which serves as a store of glucose in the body. It has separate glucosyltransferase and glucosidase activities.
Glycogen storage disease type IV (GSD IV), or Andersen's Disease, is a form of glycogen storage disease, which is caused by an inborn error of metabolism. It is the result of a mutation in the GBE1 gene, which causes a defect in the glycogen branching enzyme. Therefore, glycogen is not made properly and abnormal glycogen molecules accumulate in cells; most severely in cardiac and muscle cells. The severity of this disease varies on the amount of enzyme produced. GSD IV is autosomal recessive, which means each parent has a mutant copy of the gene, but show no symptoms of the disease. Having an autosomal recessive inheritance pattern, males and females are equally likely to be affected by Andersen's disease. Classic Andersen's disease typically becomes apparent during the first few months after the patient is born. Approximately 1 in 20,000 to 25,000 newborns have a glycogen storage disease. Andersen's disease affects 1 in 800,000 individuals worldwide, with 3% of all GSDs being type IV. The disease was described and studied first by Dorothy Hansine Andersen.
β-Amylase is an enzyme with the systematic name 4-α-D-glucan maltohydrolase. It catalyses the following reaction:
α-Glucosidase (EC 3.2.1.20, is a glucosidase located in the brush border of the small intestine that acts upon α bonds:
Phosphorylase kinase (PhK) is a serine/threonine-specific protein kinase which activates glycogen phosphorylase to release glucose-1-phosphate from glycogen. PhK phosphorylates glycogen phosphorylase at two serine residues, triggering a conformational shift which favors the more active glycogen phosphorylase “a” form over the less active glycogen phosphorylase b.
Equine polysaccharide storage myopathy is a hereditary glycogen storage disease of horses that causes exertional rhabdomyolysis. It is currently known to affect the following breeds American Quarter Horses, American Paint Horses, Warmbloods, Cobs, Dales Ponies, Thoroughbreds, Arabians, New Forest ponies, and a large number of Heavy horse breeds. While incurable, PSSM can be managed with appropriate diet and exercise. There are currently 2 subtypes, known as Type 1 PSSM and Type 2 PSSM.
Myophosphorylase or glycogen phosphorylase, muscle associated (PYGM) is the muscle isoform of the enzyme glycogen phosphorylase and is encoded by the PYGM gene. This enzyme helps break down glycogen into glucose-1-phosphate, so it can be used within the muscle cell. Mutations in this gene are associated with McArdle disease, a glycogen storage disease of muscle.
Glucan 1,4-α-glucosidase is an enzyme located on the brush border of the small intestine with systematic name 4-α-D-glucan glucohydrolase. It catalyses the following chemical reaction
In enzymology, a starch synthase is an enzyme that catalyzes the chemical reaction
Glucose-6-phosphatase, catalytic subunit is an enzyme that in humans is encoded by the G6PC gene.
α-Glucans (alpha-glucans) are polysaccharides of D-glucose monomers linked with glycosidic bonds of the alpha form. α-Glucans use cofactors in a cofactor site in order to activate a glucan phosphorylase enzyme. This enzyme causes a reaction that transfers a glucosyl portion between orthophosphate and α-I,4-glucan. The position of the cofactors to the active sites on the enzyme are critical to the overall reaction rate thus, any alteration to the cofactor site leads to the disruption of the glucan binding site.
Glucansucrase is an enzyme in the glycoside hydrolase family GH70 used by lactic acid bacteria to split sucrose and use resulting glucose molecules to build long, sticky biofilm chains. These extracellular homopolysaccharides are called α-glucan polymers.
Glucan 1,4-alpha-maltohydrolase is an enzyme with systematic name 4-alpha-D-glucan alpha-maltohydrolase. This enzyme catalyses the following chemical reaction
Neopullulanase is an enzyme of the alpha-amylase family with systematic name pullulan 4-D-glucanohydrolase (panose-forming). This enzyme principally catalyses the following chemical reaction by cleaving pullulan's alpha-1,4-glucosidic bonds:
The enzyme exo-(1→4)-α-D-glucan lyase (EC 4.2.2.13, α-(1→4)-glucan 1,5-anhydro-D-fructose eliminase, α-1,4-glucan exo-lyase, α-1,4-glucan lyase, GLase) is an enzyme with systematic name (1→4)-α-D-glucan exo-4-lyase (1,5-anhydro-D-fructose-forming). This enzyme catalyses the following chemical reaction
↑ Devillers CH, Piper ME, Ballicora MA, Preiss J (October 2003). "Characterization of the branching patterns of glycogen branching enzyme truncated on the N-terminus". Archives of Biochemistry and Biophysics. 418 (1): 34–8. doi:10.1016/S0003-9861(03)00341-2. PMID13679080.
↑ Noguchi J, Chaen K, Vu NT, Akasaka T, Shimada H, Nakashima T, Nishi A, Satoh H, Omori T, Kakuta Y, Kimura M (August 2011). "Crystal structure of the branching enzyme I (BEI) from Oryza sativa L with implications for catalysis and substrate binding". Glycobiology. 21 (8): 1108–16. doi:10.1093/glycob/cwr049. PMID21493662.
↑ Rose S (1999). The Chemistry of Life. Pelican Books. pp.199–201.
↑ Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffrè B, Donati MA, Introvini P, Alegria A, Assereto S, Morandi L, Mora M, Tonoli E, Mascelli S, Traverso M, Pasquini E, Bado M, Vilarinho L, van Noort G, Mosca F, DiMauro S, Zara F, Minetti C (September 2004). "Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV)". Neurology. 63 (6): 1053–8. doi:10.1212/01.wnl.0000138429.11433.0d. PMID15452297. S2CID7874969.
↑ Hussain A, Armistead J, Gushulak L, Kruck C, Pind S, Triggs-Raine B, Natowicz MR (September 2012). "The adult polyglucosan body disease mutation GBE1 c.1076A>C occurs at high frequency in persons of Ashkenazi Jewish background". Biochemical and Biophysical Research Communications. 426 (2): 286–8. doi:10.1016/j.bbrc.2012.08.089. PMID22943850.
Barker SA, Bourne E, Peat S (1949). "The enzymic synthesis and degradation of starch. Part IV. The purification and storage of the Q-enzyme of the potato". Journal of the Chemical Society (Resumed): 1705–1711. doi:10.1039/jr9490001705.
Hehre EJ (1951). "Enzymic Synthesis of Polysaccharides: A Biological type of Polymerization". Advances in Enzymology and Related Areas of Molecular Biology. Advances in Enzymology - and Related Areas of Molecular Biology. Vol.11. pp.297–337. doi:10.1002/9780470122563.ch6. ISBN978-0-470-12256-3. PMID24540594.{{cite book}}: |journal= ignored (help)
Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksöz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE (September 2005). "A human protein-protein interaction network: a resource for annotating the proteome". Cell. 122 (6): 957–68. doi:10.1016/j.cell.2005.08.029. hdl:11858/00-001M-0000-0010-8592-0. PMID16169070. S2CID8235923.
Massa R, Bruno C, Martorana A, de Stefano N, van Diggelen OP, Federico A (April 2008). "Adult polyglucosan body disease: proton magnetic resonance spectroscopy of the brain and novel mutation in the GBE1 gene". Muscle & Nerve. 37 (4): 530–6. doi:10.1002/mus.20916. PMID17994551. S2CID18749059.
Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffrè B, Donati MA, Introvini P, Alegria A, Assereto S, Morandi L, Mora M, Tonoli E, Mascelli S, Traverso M, Pasquini E, Bado M, Vilarinho L, van Noort G, Mosca F, DiMauro S, Zara F, Minetti C (September 2004). "Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV)". Neurology. 63 (6): 1053–8. doi:10.1212/01.wnl.0000138429.11433.0d. PMID15452297. S2CID7874969.
McCarthy JJ, Meyer J, Moliterno DJ, Newby LK, Rogers WJ, Topol EJ (December 2003). "Evidence for substantial effect modification by gender in a large-scale genetic association study of the metabolic syndrome among coronary heart disease patients". Human Genetics. 114 (1): 87–98. doi:10.1007/s00439-003-1026-1. PMID14557872. S2CID2568593.
Flachsbart F, Franke A, Kleindorp R, Caliebe A, Blanché H, Schreiber S, Nebel A (December 2010). "Investigation of genetic susceptibility factors for human longevity - a targeted nonsynonymous SNP study". Mutation Research. 694 (1–2): 13–9. doi:10.1016/j.mrfmmm.2010.08.006. PMID20800603.
Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ (January 2005). "Immunoaffinity profiling of tyrosine phosphorylation in cancer cells". Nature Biotechnology. 23 (1): 94–101. doi:10.1038/nbt1046. PMID15592455. S2CID7200157.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.