Related Research Articles
In biology, phylogenetics is the study of the evolutionary history and relationships among or within groups of organisms. These relationships are determined by phylogenetic inference methods that focus on observed heritable traits, such as DNA sequences, protein amino acid sequences, or morphology. The result of such an analysis is a phylogenetic tree—a diagram containing a hypothesis of relationships that reflects the evolutionary history of a group of organisms.
Molecular phylogenetics is the branch of phylogeny that analyzes genetic, hereditary molecular differences, predominantly in DNA sequences, to gain information on an organism's evolutionary relationships. From these analyses, it is possible to determine the processes by which diversity among species has been achieved. The result of a molecular phylogenetic analysis is expressed in a phylogenetic tree. Molecular phylogenetics is one aspect of molecular systematics, a broader term that also includes the use of molecular data in taxonomy and biogeography.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.

Excavata is an extensive and diverse but paraphyletic group of unicellular Eukaryota. The group was first suggested by Simpson and Patterson in 1999 and the name latinized and assigned a rank by Thomas Cavalier-Smith in 2002. It contains a variety of free-living and symbiotic protists, and includes some important parasites of humans such as Giardia and Trichomonas. Excavates were formerly considered to be included in the now obsolete Protista kingdom. They were distinguished from other lineages based on electron-microscopic information about how the cells are arranged. They are considered to be a basal flagellate lineage.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.
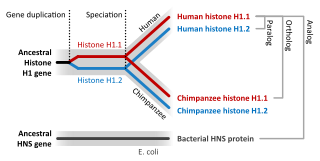
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
Computational phylogenetics, phylogeny inference, or phylogenetic inference focuses on computational and optimization algorithms, heuristics, and approaches involved in phylogenetic analyses. The goal is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of genes, species, or taxa. Maximum likelihood, parsimony, Bayesian, and minimum evolution are typical optimality criteria used to assess how well a phylogenetic tree topology describes the sequence data. Nearest Neighbour Interchange (NNI), Subtree Prune and Regraft (SPR), and Tree Bisection and Reconnection (TBR), known as tree rearrangements, are deterministic algorithms to search for optimal or the best phylogenetic tree. The space and the landscape of searching for the optimal phylogenetic tree is known as phylogeny search space.

Masatoshi Nei was a Japanese-born American evolutionary biologist.

Horizontal gene transfer (HGT) refers to the transfer of genes between distant branches on the tree of life. In evolution, it can scramble the information needed to reconstruct the phylogeny of organisms, how they are related to one another.
A conserved non-coding sequence (CNS) is a DNA sequence of noncoding DNA that is evolutionarily conserved. These sequences are of interest for their potential to regulate gene production.
PhylomeDB is a public biological database for complete catalogs of gene phylogenies (phylomes). It allows users to interactively explore the evolutionary history of genes through the visualization of phylogenetic trees and multiple sequence alignments. Moreover, phylomeDB provides genome-wide orthology and paralogy predictions which are based on the analysis of the phylogenetic trees. The automated pipeline used to reconstruct trees aims at providing a high-quality phylogenetic analysis of different genomes, including Maximum Likelihood tree inference, alignment trimming and evolutionary model testing.

The eocyte hypothesis in evolutionary biology proposes that the eukaryotes originated from a group of prokaryotes called eocytes. After his team at the University of California, Los Angeles discovered eocytes in 1984, James A. Lake formulated the hypothesis as "eocyte tree" that proposed eukaryotes as part of archaea. Lake hypothesised the tree of life as having only two primary branches: prokaryotes, which include Bacteria and Archaea, and karyotes, that comprise Eukaryotes and eocytes. Parts of this early hypothesis were revived in a newer two-domain system of biological classification which named the primary domains as Archaea and Bacteria.
Microbial phylogenetics is the study of the manner in which various groups of microorganisms are genetically related. This helps to trace their evolution. To study these relationships biologists rely on comparative genomics, as physiology and comparative anatomy are not possible methods.
Horizontal or lateral gene transfer is the transmission of portions of genomic DNA between organisms through a process decoupled from vertical inheritance. In the presence of HGT events, different fragments of the genome are the result of different evolutionary histories. This can therefore complicate investigations of the evolutionary relatedness of lineages and species. Also, as HGT can bring into genomes radically different genotypes from distant lineages, or even new genes bearing new functions, it is a major source of phenotypic innovation and a mechanism of niche adaptation. For example, of particular relevance to human health is the lateral transfer of antibiotic resistance and pathogenicity determinants, leading to the emergence of pathogenic lineages.
PICRUSt is a bioinformatics software package. The name is an abbreviation for Phylogenetic Investigation of Communities by Reconstruction of Unobserved States.
In molecular phylogenetics, relationships among individuals are determined using character traits, such as DNA, RNA or protein, which may be obtained using a variety of sequencing technologies. High-throughput next-generation sequencing has become a popular technique in transcriptomics, which represent a snapshot of gene expression. In eukaryotes, making phylogenetic inferences using RNA is complicated by alternative splicing, which produces multiple transcripts from a single gene. As such, a variety of approaches may be used to improve phylogenetic inference using transcriptomic data obtained from RNA-Seq and processed using computational phylogenetics.
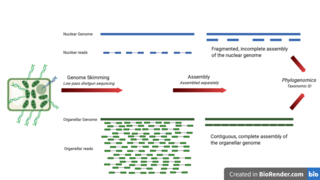
Genome skimming is a sequencing approach that uses low-pass, shallow sequencing of a genome, to generate fragments of DNA, known as genome skims. These genome skims contain information about the high-copy fraction of the genome. The high-copy fraction of the genome consists of the ribosomal DNA, plastid genome (plastome), mitochondrial genome (mitogenome), and nuclear repeats such as microsatellites and transposable elements. It employs high-throughput, next generation sequencing technology to generate these skims. Although these skims are merely 'the tip of the genomic iceberg', phylogenomic analysis of them can still provide insights on evolutionary history and biodiversity at a lower cost and larger scale than traditional methods. Due to the small amount of DNA required for genome skimming, its methodology can be applied in other fields other than genomics. Tasks like this include determining the traceability of products in the food industry, enforcing international regulations regarding biodiversity and biological resources, and forensics.
References
- ↑ BioMed Central | Fgenerated title -->
- ↑ Kumar S, Filipski AJ, Battistuzzi FU, Kosakovsky Pond SL, Tamura K (February 2012). "Statistics and truth in phylogenomics". Molecular Biology and Evolution. 29 (2): 457–472. doi:10.1093/molbev/msr202. PMC 3258035 . PMID 21873298.
- ↑ Pennisi E (June 2008). "Evolution. Building the tree of life, genome by genome". Science. 320 (5884): 1716–1717. doi:10.1126/science.320.5884.1716. PMID 18583591. S2CID 206580993.
- ↑ Simion P, Delsuc F, Phillipe H (2020). "2.1 To What Extent Current Limits of Phylogenomics Can Be Overcome?". Phylogenetics in the Genomic Era. pp. 2.1.1–2.1.34.
- 1 2 Eisen JA, Kaiser D, Myers RM (October 1997). "Gastrogenomic delights: a movable feast". Nature Medicine. 3 (10): 1076–1078. doi:10.1038/nm1097-1076. PMC 3155951 . PMID 9334711.
- ↑ Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. (August 1997). "The complete genome sequence of the gastric pathogen Helicobacter pylori". Nature. 388 (6642): 539–547. doi: 10.1038/41483 . PMID 9252185.
- ↑ Eisen JA (March 1998). "Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis". Genome Research. 8 (3): 163–167. doi: 10.1101/gr.8.3.163 . PMID 9521918.
- ↑ Brown D, Sjölander K (June 2006). "Functional classification using phylogenomic inference". PLOS Computational Biology. 2 (6): e77. Bibcode:2006PLSCB...2...77B. doi: 10.1371/journal.pcbi.0020077 . PMC 1484587 . PMID 16846248.
- ↑ Sjölander K (January 2004). "Phylogenomic inference of protein molecular function: advances and challenges". Bioinformatics. 20 (2): 170–179. doi: 10.1093/bioinformatics/bth021 . PMID 14734307.
- ↑ Whitaker JW, McConkey GA, Westhead DR (2009). "The transferome of metabolic genes explored: analysis of the horizontal transfer of enzyme encoding genes in unicellular eukaryotes". Genome Biology. 10 (4): R36. doi: 10.1186/gb-2009-10-4-r36 . PMC 2688927 . PMID 19368726.
- ↑ Delsuc F, Brinkmann H, Philippe H (May 2005). "Phylogenomics and the reconstruction of the tree of life". Nature Reviews. Genetics. 6 (5): 361–375. CiteSeerX 10.1.1.333.1615 . doi:10.1038/nrg1603. PMID 15861208. S2CID 16379422.
- ↑ Philippe H, Snell EA, Bapteste E, Lopez P, Holland PW, Casane D "Phylogenomics of eukaryotes: impact of missing data on large alignments Mol Biol Evol 2004 Sep;21(9):1740-52. .
- ↑ Jeffroy O, Brinkmann H, Delsuc F, Philippe H (April 2006). "Phylogenomics: the beginning of incongruence?" (PDF). Trends in Genetics. 22 (4): 225–231. doi:10.1016/j.tig.2006.02.003. PMID 16490279.
- ↑ Burki F, Shalchian-Tabrizi K, Pawlowski J (August 2008). "Phylogenomics reveals a new 'megagroup' including most photosynthetic eukaryotes". Biology Letters. 4 (4): 366–369. doi:10.1098/rsbl.2008.0224. PMC 2610160 . PMID 18522922.
- ↑ dos Reis M, Inoue J, Hasegawa M, Asher RJ, Donoghue PC, Yang Z (September 2012). "Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny". Proceedings. Biological Sciences. 279 (1742): 3491–3500. doi:10.1098/rspb.2012.0683. PMC 3396900 . PMID 22628470.
- ↑ Kober KM, Bernardi G (April 2013). "Phylogenomics of strongylocentrotid sea urchins". BMC Evolutionary Biology. 13: 88. doi: 10.1186/1471-2148-13-88 . PMC 3637829 . PMID 23617542.
- ↑ Philippe, Herve'; Delsuc, Frederic; Brinkmann, Henner; Lartillot, Nicolas (2005). "Phylogenomics". Annual Review of Ecology, Evolution, and Systematics. 36: 541–562. doi:10.1146/annurev.ecolsys.35.112202.130205.
| Relevant fields | ||
|---|---|---|
| Basic concepts | ||
| Inference methods | ||
| Current topics | ||
| Group traits | ||
| Group types | ||
| Nomenclature | ||