A secondary sex characteristic is a physical characteristic of an organism that is related to or derived from its sex, but not directly part of its reproductive system. In humans, these characteristics typically start to appear during puberty. In animals, they can start to appear at sexual maturity. In humans, secondary sex characteristics include enlarged breasts and widened hips of females, facial hair and Adam's apples on males, and pubic hair on both. In non-human animals, secondary sex characteristics include, for example, the manes of male lions, the bright facial and rump coloration of male mandrills, and horns in many goats and antelopes.
An androgen is any natural or synthetic steroid hormone that regulates the development and maintenance of male characteristics in vertebrates by binding to androgen receptors. This includes the embryological development of the primary male sex organs, and the development of male secondary sex characteristics at puberty. Androgens are synthesized in the testes, the ovaries, and the adrenal glands.

Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. It results from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. Most of these disorders involve excessive or deficient production of hormones such as glucocorticoids, mineralocorticoids, or sex steroids, and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults. It is one of the most common autosomal recessive disorders in humans.

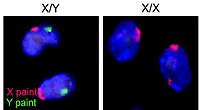
XY complete gonadal dysgenesis, also known as Swyer syndrome, is a type of defect hypogonadism in a person whose karyotype is 46,XY. Though they typically have normal vulvas, the person has underdeveloped gonads, fibrous tissue termed "streak gonads", and if left untreated, will not experience puberty. The cause is a lack or inactivation of an SRY gene which is responsible for sexual differentiation. Pregnancy is often possible in Swyer syndrome with assisted reproductive technology. The phenotype is usually similar to Turner syndrome (45,X0) due to a lack of X inactivation. The typical medical treatment is hormone replacement therapy. The syndrome was named after Gerald Swyer, an endocrinologist based in London.

Lipoid congenital adrenal hyperplasia is an endocrine disorder that is an uncommon and potentially lethal form of congenital adrenal hyperplasia (CAH). It arises from defects in the earliest stages of steroid hormone synthesis: the transport of cholesterol into the mitochondria and the conversion of cholesterol to pregnenolone—the first step in the synthesis of all steroid hormones. Lipoid CAH causes mineralocorticoid deficiency in affected infants and children. Male infants are severely undervirilized causing their external genitalia to look feminine. The adrenals are large and filled with lipid globules derived from cholesterol.
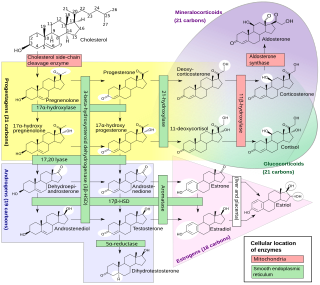
Congenital adrenal hyperplasia due to 11β-hydroxylase deficiency is a form of congenital adrenal hyperplasia (CAH) which produces a higher than normal amount of androgen, resulting from a defect in the gene encoding the enzyme steroid 11β-hydroxylase (11β-OH) which mediates the final step of cortisol synthesis in the adrenal. 11β-OH CAH results in hypertension due to excessive mineralocorticoid effects. It also causes excessive androgen production both before and after birth and can virilize a genetically female fetus or a child of either sex.

Congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene for one of the key enzymes in cortisol synthesis by the adrenal gland, 3β-hydroxysteroid dehydrogenase (3β-HSD) type II (HSD3B2). As a result, higher levels of 17α-hydroxypregnenolone appear in the blood with adrenocorticotropic hormone (ACTH) challenge, which stimulates adrenal corticosteroid synthesis.
Congenital adrenal hyperplasia due to 17α-hydroxylase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene CYP17A1, which produces the enzyme 17α-hydroxylase. It causes decreased synthesis of cortisol and sex hormones, with resulting increase in mineralocorticoid production. Thus, common symptoms include mild cortisol deficiency, ambiguous genitalia in men or amenorrhea at puberty in women, and hypokalemic hypertension. However, partial (incomplete) deficiency often has inconsistent symptoms between patients, and affected women may be asymptomatic except for infertility.
Pubarche refers to the first appearance of pubic hair at puberty and it also marks the beginning of puberty. It is one of the physical changes of puberty and can occur independently of complete puberty. The early stage of sexual maturation, also known as adrenarche, is marked by characteristics including the development of pubic hair, axillary hair, adult apocrine body odor, acne, and increased oiliness of hair and skin. The Encyclopedia of Child and Adolescent Health corresponds SMR2 with pubarche, defining it as the development of pubic hair that occurs at a mean age of 11.6 years in females and 12.6 years in males. It further describes that pubarche's physical manifestation is vellus hair over the labia or the base of the penis. See Table 1 for the entirety of the sexual maturity rating description.

Intersex medical interventions (IMI), sometimes known as intersex genital mutilations (IGM), are surgical, hormonal and other medical interventions performed to modify atypical or ambiguous genitalia and other sex characteristics, primarily for the purposes of making a person's appearance more typical and to reduce the likelihood of future problems. The history of intersex surgery has been characterized by controversy due to reports that surgery can compromise sexual function and sensation, and create lifelong health issues. The medical interventions can be for a variety of reasons, due to the enormous variety of the disorders of sex development. Some disorders, such as salt-wasting disorder, can be life-threatening if left untreated.

Sex-determining region Y protein (SRY), or testis-determining factor (TDF), is a DNA-binding protein encoded by the SRY gene that is responsible for the initiation of male sex determination in therian mammals. SRY is an intronless sex-determining gene on the Y chromosome. Mutations in this gene lead to a range of disorders of sex development with varying effects on an individual's phenotype and genotype.

The male reproductive system consists of a number of sex organs that play a role in the process of human reproduction. These organs are located on the outside of the body, and within the pelvis.

XX male syndrome, also known as de la Chapelle syndrome, is a rare condition in which an individual with a 46,XX karyotype develops a male phenotype. Synonyms for XX male syndrome include 46,XX testicular difference of sex development
Gonadal dysgenesis is classified as any congenital developmental disorder of the reproductive system in humans. It is atypical development of gonads in an embryo. One type of gonadal dysgenesis is the development of functionless, fibrous tissue, termed streak gonads, instead of reproductive tissue. Streak gonads are a form of aplasia, resulting in hormonal failure that manifests as sexual infantism and infertility, with no initiation of puberty and secondary sex characteristics.

Partial androgen insensitivity syndrome (PAIS) is a condition that results in the partial inability of the cell to respond to androgens. It is an X linked recessive condition. The partial unresponsiveness of the cell to the presence of androgenic hormones impairs the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does not significantly impair female genital or sexual development. As such, the insensitivity to androgens is clinically significant only when it occurs in individuals with a Y chromosome. Clinical features include ambiguous genitalia at birth and primary amenhorrhoea with clitoromegaly with inguinal masses. Müllerian structures are not present in the individual.

Sexual differentiation in humans is the process of development of sex differences in humans. It is defined as the development of phenotypic structures consequent to the action of hormones produced following gonadal determination. Sexual differentiation includes development of different genitalia and the internal genital tracts and body hair plays a role in sex identification.
Disorders of sex development (DSDs), also known as differences in sex development or variations in sex characteristics (VSC), are congenital conditions affecting the reproductive system, in which development of chromosomal, gonadal, or anatomical sex is atypical.
Puberty is the process of physical changes through which a child's body matures into an adult body capable of sexual reproduction. It is initiated by hormonal signals from the brain to the gonads: the ovaries in a female, the testicles in a male. In response to the signals, the gonads produce hormones that stimulate libido and the growth, function, and transformation of the brain, bones, muscle, blood, skin, hair, breasts, and sex organs. Physical growth—height and weight—accelerates in the first half of puberty and is completed when an adult body has been developed. Before puberty, the external sex organs, known as primary sexual characteristics, are sex characteristics that distinguish males and females. Puberty leads to sexual dimorphism through the development of the secondary sex characteristics, which further distinguish the sexes.
Sexual anomalies, also known as sexual abnormalities, are a set of clinical conditions due to chromosomal, gonadal and/or genitalia variation. Individuals with congenital (inborn) discrepancy between sex chromosome, gonadal, and their internal and external genitalia are categorised as individuals with a disorder of sex development (DSD). Afterwards, if the family or individual wishes, they can partake in different management and treatment options for their conditions.
Various criteria have been offered for the definition of intersex, including ambiguous genitalia, atypical genitalia, and differential sexual development. Ambiguous genitalia occurs in roughly 0.05% of all births, and atypical genitalia occurs in 0.5% of all births, usually caused by masculinization or feminization during pregnancy, these conditions range from full androgen insensitivity syndrome to ovotesticular syndrome, although the definition of what constitutes "normal" genitalia is largely arbitrary.