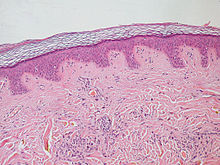
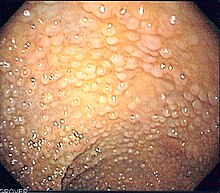
A lipoma is a benign tumor made of fat tissue. They are generally soft to the touch, movable, and painless. They usually occur just under the skin, but occasionally may be deeper. Most are less than 5 cm (2.0 in) in size. Common locations include upper back, shoulders, and abdomen. It is possible to have a number of lipomas.

Proteus syndrome is a rare disorder with a genetic background that can cause tissue overgrowth involving all three embryonic lineages. Patients with Proteus syndrome tend to have an increased risk of embryonic tumor development. The clinical and radiographic symptoms of Proteus syndrome are highly variable, as are its orthopedic manifestations.

Malignancy is the tendency of a medical condition to become progressively worse; the term is most familiar as a characterization of cancer.

Pituitary adenomas are tumors that occur in the pituitary gland. Most pituitary tumors are benign, approximately 35% are invasive and just 0.1% to 0.2% are carcinomas. Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms and the estimated prevalence rate in the general population is approximately 17%.

A neoplasm is a type of abnormal and excessive growth of tissue. The process that occurs to form or produce a neoplasm is called neoplasia. The growth of a neoplasm is uncoordinated with that of the normal surrounding tissue, and persists in growing abnormally, even if the original trigger is removed. This abnormal growth usually forms a mass, when it may be called a tumour or tumor.

Enchondroma is a type of benign bone tumor belonging to the group of cartilage tumors. There may be no symptoms, or it may present typically in the short tubular bones of the hands with a swelling, pain or pathological fracture.

A hamartoma is a mostly benign, local malformation of cells that resembles a neoplasm of local tissue but is usually due to an overgrowth of multiple aberrant cells, with a basis in a systemic genetic condition, rather than a growth descended from a single mutated cell (monoclonality), as would typically define a benign neoplasm/tumor. Despite this, many hamartomas are found to have clonal chromosomal aberrations that are acquired through somatic mutations, and on this basis the term hamartoma is sometimes considered synonymous with neoplasm. Hamartomas are by definition benign, slow-growing or self-limiting, though the underlying condition may still predispose the individual towards malignancies.

Osteochondromas are the most common benign tumors of the bones. The tumors take the form of cartilage-capped bony projections or outgrowth on the surface of bones exostoses. It is characterized as a type of overgrowth that can occur in any bone where cartilage forms bone. Tumors most commonly affect long bones about the knee and in the forearm. Additionally, flat bones such as the pelvis and scapula may be affected. Hereditary multiple exostoses usually present during childhood. Yet, the vast majority of affected individuals become clinically manifest by the time they reach adolescence. Osteochondromas occur in 3% of the general population and represent 35% of all benign tumors and 8% of all bone tumors. The majority of these tumors are solitary non-hereditary lesions and approximately 15% of osteochondromas occur as hereditary multiple exostoses preferably known as hereditary multiple osteochondromas (HMOs). Osteochondromas do not result from injury and the exact cause remains unknown. Recent research has indicated that multiple osteochondromas is an autosomal dominant inherited disease. Germ line mutations in EXT1 and EXT2 genes located on chromosomes 8 and 11 have been associated with the cause of the disease. The treatment choice for osteochondroma is surgical removal of solitary lesion or partial excision of the outgrowth, when symptoms cause motion limitations or nerve and blood vessel impingements. In hereditary multiple exostoses the indications of surgery are based upon multiple factors that are taken collectively, namely: patient's age, tumor location and number, accompanying symptomatology, esthetic concerns, family history and underlying gene mutation. A variety of surgical procedures have been employed to remedy hereditary multiple exostoses such as osteochondroma excision, bone lengthening, corrective osteotomy and hemiepiphysiodesis. Sometimes a combination of the previous procedures is used. The indicators of surgical success in regard to disease and patient characteristics are greatly disputable. Because most studies of hereditary multiple exostoses are retrospective and of limited sample size with missing data, the best evidence for each of the currently practiced surgical procedures is lacking.

Multiple endocrine neoplasia type 1 (MEN-1) is one of a group of disorders, the multiple endocrine neoplasias, that affect the endocrine system through development of neoplastic lesions in pituitary, parathyroid gland and pancreas. Individuals suffering from this disorder are prone to developing multiple endocrine and nonendocrine tumors. It was first described by Paul Wermer in 1954.

Cowden syndrome is an autosomal dominant inherited condition characterized by benign overgrowths called hamartomas as well as an increased lifetime risk of breast, thyroid, uterine, and other cancers. It is often underdiagnosed due to variability in disease presentation, but 99% of patients report mucocutaneous symptoms by age 20–29. Despite some considering it a primarily dermatologic condition, Cowden's syndrome is a multi-system disorder that also includes neurodevelopmental disorders such as macrocephaly.
Metachondromatosis is an autosomal dominant, incompletely penetrant genetic disease affecting the growth of bones, leading to exostoses primarily in the hands and feet as well as enchondromas of long bone metaphyses and iliac crests. This syndrome affects mainly tubular bones, though it can also involve the vertebrae, small joints, and flat bones. The disease is thought to affect exon 4 of the PTPN11 gene. Metachondromatosis is believed to be caused by an 11 base pair deletion resulting in a frameshift and nonsense mutation. The disease was discovered and named in 1971 by Pierre Maroteaux, a French physician, when he observed two families with skeletal radiologic features with exostoses and Ollier disease. The observation of one family with five affected people led to the identification of the disease as autosomal dominant. There have been less than 40 cases of the disease reported to date.
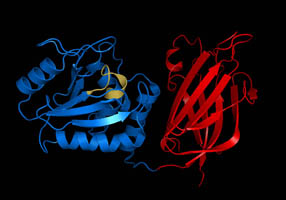
Phosphatase and tensin homolog (PTEN) is a phosphatase in humans and is encoded by the PTEN gene. Mutations of this gene are a step in the development of many cancers, specifically glioblastoma, lung cancer, breast cancer, and prostate cancer. Genes corresponding to PTEN (orthologs) have been identified in most mammals for which complete genome data are available.
Bannayan–Riley–Ruvalcaba syndrome (BRRS) is a rare overgrowth syndrome and hamartomatous disorder with occurrence of multiple subcutaneous lipomas, macrocephaly and hemangiomas. The disease is inherited in an autosomal dominant manner. The disease belongs to a family of hamartomatous polyposis syndromes, which also includes Peutz–Jeghers syndrome, juvenile polyposis and Cowden syndrome. Mutation of the PTEN gene underlies this syndrome, as well as Cowden syndrome, Proteus syndrome, and Proteus-like syndrome, these four syndromes are referred to as PTEN Hamartoma-Tumor Syndromes.

Ollier disease is a rare sporadic nonhereditary skeletal disorder in which typically benign cartilaginous tumors (enchondromas) develop near the growth plate cartilage. This is caused by cartilage rests that grow and reside within the metaphysis or diaphysis and eventually mineralize over time to form multiple enchondromas. Key signs of the disorder include asymmetry and shortening of the limb as well as an increased thickness of the bone margin. These symptoms are typically first visible during early childhood with the mean age of diagnosis being 13 years of age. Many patients with Ollier disease are prone to develop other malignancies including bone sarcomas that necessitate treatment and the removal of malignant bone neoplasm. Cases in patients with Ollier disease has shown a link to IDH1, IDH2, and PTH1R gene mutations. Currently, there are no forms of treatment for the underlying condition of Ollier disease but complications such as fractures, deformities, malignancies that arise from it can be treated through surgical procedures. The prevalence of this condition is estimated at around 1 in 100,000. It is unclear whether the men or women are more affected by this disorder due to conflicting case studies.
Maffucci syndrome is a very rare disorder in which multiple benign tumors of cartilage develop within the bones. The tumors most commonly appear in the bones of the hands, feet, and limbs, causing bone deformities and short limbs.

Endometrial intraepithelial neoplasia (EIN) is a premalignant lesion of the uterine lining that predisposes to endometrioid endometrial adenocarcinoma. It is composed of a collection of abnormal endometrial cells, arising from the glands that line the uterus, which have a tendency over time to progress to the most common form of uterine cancer—endometrial adenocarcinoma, endometrioid type.

Juvenile polyposis syndrome is an autosomal dominant genetic condition characterized by the appearance of multiple juvenile polyps in the gastrointestinal tract. Polyps are abnormal growths arising from a mucous membrane. These usually begin appearing before age 20, but the term juvenile refers to the type of polyp, not to the age of the affected person. While the majority of the polyps found in juvenile polyposis syndrome are non-neoplastic, hamartomatous, self-limiting and benign, there is an increased risk of adenocarcinoma.

A colorectal polyp is a polyp occurring on the lining of the colon or rectum. Untreated colorectal polyps can develop into colorectal cancer.
Vulvar tumors are those neoplasms of the vulva. Vulvar and vaginal neoplasms make up a small percentage (3%) of female genital cancers. They can be benign or malignant. Vulvar neoplasms are divided into cystic or solid lesions and other mixed types. Vulvar cancers are those malignant neoplasms that originate from vulvar epithelium, while vulvar sarcomas develop from non-epithelial cells such as bone, cartilage, fat, muscle, blood vessels, or other connective or supportive tissue. Epithelial and mesenchymal tissue are the origin of vulvar tumors.
CYLD cutaneous syndrome (CCS) is the recently designated term for three rare inherited cutaneous adnexal tumor syndromes: multiple familial trichoepithelioma (MFT1), Brooke–Spiegler syndrome (BSS), and familial cylindromatosis (FC). Cutaneous adnexal tumors are a large group of skin tumors that consist of tissues that have differentiated towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands. CCS tumors are hair follicle tumors.