Histone acetyltransferases (HATs) are enzymes that acetylate conserved lysine amino acids on histone proteins by transferring an acetyl group from acetyl-CoA to form ε-N-acetyllysine. DNA is wrapped around histones, and, by transferring an acetyl group to the histones, genes can be turned on and off. In general, histone acetylation increases gene expression.

Histone deacetylases (EC 3.5.1.98, HDAC) are a class of enzymes that remove acetyl groups (O=C-CH3) from an ε-N-acetyl lysine amino acid on both histone and non-histone proteins. HDACs allow histones to wrap the DNA more tightly. This is important because DNA is wrapped around histones, and DNA expression is regulated by acetylation and de-acetylation. HDAC's action is opposite to that of histone acetyltransferase. HDAC proteins are now also called lysine deacetylases (KDAC), to describe their function rather than their target, which also includes non-histone proteins. In general, they suppress gene expression.
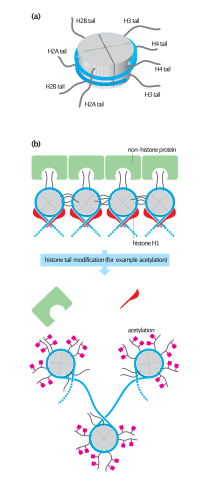
The histone code is a hypothesis that the transcription of genetic information encoded in DNA is in part regulated by chemical modifications to histone proteins, primarily on their unstructured ends. Together with similar modifications such as DNA methylation it is part of the epigenetic code. Histones associate with DNA to form nucleosomes, which themselves bundle to form chromatin fibers, which in turn make up the more familiar chromosome. Histones are globular proteins with a flexible N-terminus that protrudes from the nucleosome. Many of the histone tail modifications correlate very well to chromatin structure and both histone modification state and chromatin structure correlate well to gene expression levels. The critical concept of the histone code hypothesis is that the histone modifications serve to recruit other proteins by specific recognition of the modified histone via protein domains specialized for such purposes, rather than through simply stabilizing or destabilizing the interaction between histone and the underlying DNA. These recruited proteins then act to alter chromatin structure actively or to promote transcription. For details of gene expression regulation by histone modifications see table below.

Acetyllysine is an acetyl-derivative of the amino acid lysine. There are multiple forms of acetyllysine: this article is about N-ε-acetyl-L-lysine; the other form is N-α-acetyl-L-lysine.
Histone deacetylase inhibitors are chemical compounds that inhibit histone deacetylases. Since deacetylation of histones produces transcriptionally silenced euchromatin, HDIs can render chromatin more transcriptionally active and induce epigenomic changes.
Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. Such remodeling is principally carried out by 1) covalent histone modifications by specific enzymes, e.g., histone acetyltransferases (HATs), deacetylases, methyltransferases, and kinases, and 2) ATP-dependent chromatin remodeling complexes which either move, eject or restructure nucleosomes. Besides actively regulating gene expression, dynamic remodeling of chromatin imparts an epigenetic regulatory role in several key biological processes, egg cells DNA replication and repair; apoptosis; chromosome segregation as well as development and pluripotency. Aberrations in chromatin remodeling proteins are found to be associated with human diseases, including cancer. Targeting chromatin remodeling pathways is currently evolving as a major therapeutic strategy in the treatment of several cancers.
While the cellular and molecular mechanisms of learning and memory have long been a central focus of neuroscience, it is only in recent years that attention has turned to the epigenetic mechanisms behind the dynamic changes in gene transcription responsible for memory formation and maintenance. Epigenetic gene regulation often involves the physical marking of DNA or associated proteins to cause or allow long-lasting changes in gene activity. Epigenetic mechanisms such as DNA methylation and histone modifications have been shown to play an important role in learning and memory.
Epigenetic regulation of neurogenesis is the role that epigenetics plays in the regulation of neurogenesis.
H3K27ac is an epigenetic modification to the DNA packaging protein histone H3. It is a mark that indicates acetylation of the lysine residue at N-terminal position 27 of the histone H3 protein.
H2BK5ac is an epigenetic modification to the DNA packaging protein Histone H2B. It is a mark that indicates the acetylation at the 5th lysine residue of the histone H2B protein. H2BK5ac is involved in maintaining stem cells and colon cancer.
H4K16ac is an epigenetic modification to the DNA packaging protein Histone H4. It is a mark that indicates the acetylation at the 16th lysine residue of the histone H4 protein.
H4K5ac is an epigenetic modification to the DNA packaging protein histone H4. It is a mark that indicates the acetylation at the 5th lysine residue of the histone H4 protein. H4K5 is the closest lysine residue to the N-terminal tail of histone H4. It is enriched at the transcription start site (TSS) and along gene bodies. Acetylation of histone H4K5 and H4K12ac is enriched at centromeres.
H4K8ac, representing an epigenetic modification to the DNA packaging protein histone H4, is a mark indicating the acetylation at the 8th lysine residue of the histone H4 protein. It has been implicated in the prevalence of malaria.
H4K12ac is an epigenetic modification to the DNA packaging protein histone H4. It is a mark that indicates the acetylation at the 12th lysine residue of the histone H4 protein. H4K12ac is involved in learning and memory. It is possible that restoring this modification could reduce age-related decline in memory.
H4K91ac is an epigenetic modification to the DNA packaging protein histone H4. It is a mark that indicates the acetylation at the 91st lysine residue of the histone H4 protein. No known diseases are attributed to this mark but it might be implicated in melanoma.
H3K23ac is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the acetylation at the 23rd lysine residue of the histone H3 protein.
H3K14ac is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the acetylation at the 14th lysine residue of the histone H3 protein.
H3K9ac is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the acetylation at the 9th lysine residue of the histone H3 protein.
H3K36ac is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the acetylation at the 36th lysine residue of the histone H3 protein.
H3K56ac is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the acetylation at the 56th lysine residue of the histone H3 protein.