
Bone healing, or fracture healing, is a proliferative physiological process in which the body facilitates the repair of a bone fracture.

A bone tumor is an abnormal growth of tissue in bone, traditionally classified as noncancerous (benign) or cancerous (malignant). Cancerous bone tumors usually originate from a cancer in another part of the body such as from lung, breast, thyroid, kidney and prostate. There may be a lump, pain, or neurological signs from pressure. A bone tumor might present with a pathologic fracture. Other symptoms may include fatigue, fever, weight loss, anemia and nausea. Sometimes there are no symptoms and the tumour is found when investigating another problem.

The term fibromatosis refers to a group of soft tissue tumors which have certain characteristics in common, including absence of cytologic and clinical malignant features, a histology consistent with proliferation of well-differentiated fibroblasts, an infiltrative growth pattern, and aggressive clinical behavior with frequent local recurrence. It is classed by the World Health Organization as an intermediate soft tissue tumor related to the sarcoma family. Arthur Purdy Stout coined the term fibromatosis, in 1954.
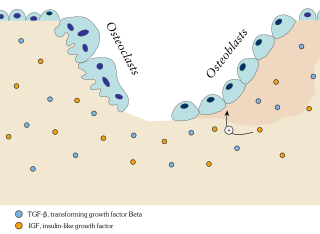
Ossification in bone remodeling is the process of laying down new bone material by cells named osteoblasts. It is synonymous with bone tissue formation. There are two processes resulting in the formation of normal, healthy bone tissue: Intramembranous ossification is the direct laying down of bone into the primitive connective tissue (mesenchyme), while endochondral ossification involves cartilage as a precursor.

Fibrodysplasia ossificans progressiva, also called Münchmeyer disease or formerly myositis ossificans progressiva, is an extremely rare connective tissue disease in which fibrous connective tissue such as muscle, tendons, and ligaments turn into bone tissue. It is the only known medical condition where one organ system changes into another. It is a severe, disabling disorder with no cure or treatment.

Calcification is the accumulation of calcium salts in a body tissue. It normally occurs in the formation of bone, but calcium can be deposited abnormally in soft tissue, causing it to harden. Calcifications may be classified on whether there is mineral balance or not, and the location of the calcification. Calcification may also refer to the processes of normal mineral deposition in biological systems, such as the formation of stromatolites or mollusc shells.

Osteochondromas are the most common benign tumors of the bones. The tumors take the form of cartilage-capped bony projections or outgrowth on the surface of bones exostoses. It is characterized as a type of overgrowth that can occur in any bone where cartilage forms bone. Tumors most commonly affect long bones about the knee and in the forearm. Additionally, flat bones such as the pelvis and scapula may be affected. Hereditary multiple exostoses usually present during childhood. Yet, the vast majority of affected individuals become clinically manifest by the time they reach adolescence. Osteochondromas occur in 3% of the general population and represent 35% of all benign tumors and 8% of all bone tumors. The majority of these tumors are solitary non-hereditary lesions and approximately 15% of osteochondromas occur as hereditary multiple exostoses preferably known as hereditary multiple osteochondromas (HMOs). Osteochondromas do not result from injury and the exact cause remains unknown. Recent research has indicated that multiple osteochondromas is an autosomal dominant inherited disease. Germ line mutations in EXT1 and EXT2 genes located on chromosomes 8 and 11 have been associated with the cause of the disease. The treatment choice for osteochondroma is surgical removal of solitary lesion or partial excision of the outgrowth, when symptoms cause motion limitations or nerve and blood vessel impingements. In hereditary multiple exostoses the indications of surgery are based upon multiple factors that are taken collectively, namely: patient's age, tumor location and number, accompanying symptomatology, esthetic concerns, family history and underlying gene mutation. A variety of surgical procedures have been employed to remedy hereditary multiple exostoses such as osteochondroma excision, bone lengthening, corrective osteotomy and hemiepiphysiodesis. Sometimes a combination of the previous procedures is used. The indicators of surgical success in regard to disease and patient characteristics are greatly disputable. Because most studies of hereditary multiple exostoses are retrospective and of limited sample size with missing data, the best evidence for each of the currently practiced surgical procedures is lacking.
Heterotopic ossification (HO) is the process by which bone tissue forms outside of the skeleton in muscles and soft tissue.

Ollier disease is a rare sporadic nonhereditary skeletal disorder in which typically benign cartilaginous tumors (enchondromas) develop near the growth plate cartilage. This is caused by cartilage rests that grow and reside within the metaphysis or diaphysis and eventually mineralize over time to form multiple enchondromas. Key signs of the disorder include asymmetry and shortening of the limb as well as an increased thickness of the bone margin. These symptoms are typically first visible during early childhood with the mean age of diagnosis being 13 years of age. Many patients with Ollier disease are prone to develop other malignancies including bone sarcomas that necessitate treatment and the removal of malignant bone neoplasm. Cases in patients with Ollier disease has shown a link to IDH1, IDH2, and PTH1R gene mutations. Currently, there are no forms of treatment for the underlying condition of Ollier disease but complications such as fractures, deformities, malignancies that arise from it can be treated through surgical procedures. The prevalence of this condition is estimated at around 1 in 100,000. It is unclear whether the men or women are more affected by this disorder due to conflicting case studies.
The calcifying epithelial odontogenic tumor (CEOT), also known as a Pindborg tumor, is an odontogenic tumor first recognized by the Danish pathologist Jens Jørgen Pindborg in 1955. It was previously described as an adenoid adamantoblastoma, unusual ameloblastoma and a cystic odontoma. Like other odontogenic neoplasms, it is thought to arise from the epithelial element of the enamel origin. It is a typically benign and slow growing, but invasive neoplasm.

Chondroblastoma is a rare, benign, locally aggressive bone tumor that typically affects the epiphyses or apophyses of long bones. It is thought to arise from an outgrowth of immature cartilage cells (chondroblasts) from secondary ossification centers, originating from the epiphyseal plate or some remnant of it.

Ectopic calcification is a pathologic deposition of calcium salts in tissues or bone growth in soft tissues. This can be a symptom of hyperphosphatemia. Formation of osseous tissue in soft tissues such as the lungs, eyes, arteries, or other organs is known as ectopic calcification, dystrophic calcification, or ectopic ossification.
Diffuse infantile fibromatosis is a rare condition affecting infants during the first three years of life. This condition is a multicentric infiltration of muscle fibers with fibroblasts resembling those seen in aponeurotic fibromas, presenting as lesions and tumors confined usually to the muscles of the arms, neck, and shoulder area Diffuse infantile fibromatosis is characterized by fast growing benign tumors. This disorder is known to be caused by mutations in germline variants, PDGFRB and NOTCH3, which may be generationally-inherited through autosomal dominant and recessive traits. Although diffuse infantile fibromatosis is classified as benign, it can still lead to life-threatening complications and damage other organs.
Harry Raymond Eastlack, Jr. was the subject of the most recognized case of FOP from the 20th century. His case is also particularly acknowledged, by scientists and researchers, for his contribution to medical advancement. After suffering from a rare, disabling, and currently incurable genetic disease, Eastlack decided to have his skeleton and medical history donated to the Mütter Museum of the College of Physicians of Philadelphia in support of FOP research. His skeleton is one of the few FOP-presenting, fully articulated ones in existence, and it has proved valuable to the study of the disease.

Diffuse idiopathic skeletal hyperostosis (DISH) is a condition characterized by abnormal calcification/bone formation (hyperostosis) of the soft tissues surrounding the joints of the spine, and also of the peripheral or appendicular skeleton. In the spine, there is bone formation along the anterior longitudinal ligament and sometimes the posterior longitudinal ligament, which may lead to partial or complete fusion of adjacent vertebrae. The facet and sacroiliac joints tend to be uninvolved. The thoracic spine is the most common level involved. In the peripheral skeleton, DISH manifests as a calcific enthesopathy, with pathologic bone formation at sites where ligaments and tendons attach to bone.
Progressive osseous heteroplasia is a cutaneous condition characterized by cutaneous or subcutaneous ossification.

Palovarotene, sold under the brand name Sohonos, is a medication used for the treatment of heterotopic ossification and fibrodysplasia ossificans progressiva. It is a highly selective retinoic acid receptor gamma (RARγ) agonist.

Eileen M. Shore is an American medical researcher and geneticist specializing in research of muscoskeletal disorders such as fibrodysplasia ossificans progressiva.

Proliferative fasciitis and proliferative myositis (PF/PM) are rare benign soft tissue lesions that increase in size over several weeks and often regress over the ensuing 1–3 months. The lesions in PF/PM are typically obvious tumors or swellings. Historically, many studies had grouped the two descriptive forms of PF/PM as similar disorders with the exception that proliferative fasciitis occurs in subcutaneous tissues while proliferative myositis occurs in muscle tissues. In 2020, the World Health Organization agreed with this view and defined these lesions as virtually identical disorders termed proliferative fasciitis/proliferative myositis or proliferative fasciitis and proliferative myositis. The Organization also classified them as one of the various forms of the fibroblastic and myofibroblastic tumors.
Fibroblastic and myofibroblastic tumors (FMTs) develop from the mesenchymal stem cells which differentiate into fibroblasts and/or the myocytes/myoblasts that differentiate into muscle cells. FMTs are a heterogeneous group of soft tissue neoplasms. The World Health Organization (2020) defined tumors as being FMTs based on their morphology and, more importantly, newly discovered abnormalities in the expression levels of key gene products made by these tumors' neoplastic cells. Histopathologically, FMTs consist of neoplastic connective tissue cells which have differented into cells that have microscopic appearances resembling fibroblasts and/or myofibroblasts. The fibroblastic cells are characterized as spindle-shaped cells with inconspicuous nucleoli that express vimentin, an intracellular protein typically found in mesenchymal cells, and CD34, a cell surface membrane glycoprotein. Myofibroblastic cells are plumper with more abundant cytoplasm and more prominent nucleoli; they express smooth muscle marker proteins such as smooth muscle actins, desmin, and caldesmon. The World Health Organization further classified FMTs into four tumor forms based on their varying levels of aggressiveness: benign, intermediate, intermediate, and malignant.
