Metabolic myopathies are myopathies that result from defects in biochemical metabolism that primarily affect muscle. They are generally genetic defects (inborn errors of metabolism) that interfere with the ability to create energy, causing a low ATP reservoir within the muscle cell.[1][2]
At the cellular level, metabolic myopathies lack some kind of enzyme or transport protein that prevents the chemical reactions necessary to create adenosine triphosphate (ATP).[1][3] ATP is often referred to as the "molecular unit of currency" of intracellular energy transfer. The lack of ATP prevents the muscle cells from being able to function properly. Some people with a metabolic myopathy never develop symptoms due to the body's ability to produce enough ATP through alternative pathways (e.g. the majority of those with AMP-deaminase deficiency are asymptomatic[1][4]).
H2O + ATP → H+ + ADP + Pi + energy → muscle contraction[5]
Firstly, ATP is needed for transport proteins to actively transportcalcium ions into the sarcoplasmic reticulum (SR) of the muscle cell between muscle contractions. Afterwards, when a nerve signal is received, calcium channels in the SR open briefly and calcium rushes into the cytosol by selective diffusion (which does not use ATP) in what is called a "calcium spark." The diffusion of calcium ions into the cytosol causes the myosin strands of the myofibril to become exposed, and the myosin strands pull the actin microfilaments together. The muscle begins to contract.[6]
Secondly, ATP is needed to allow the myosin to release and pull again, so that the muscle can contract further in what is known as the sliding filament model.[6]
Skeletal muscle contraction
ATP is consumed at a high rate by contracting muscles. The need for ATP in muscle cells is illustrated by the phenomenon of Rigor mortis, which is the muscle rigidity that occurs in dead bodies for a short time after death. In these muscles, all the ATP has been used up and in the absence of further ATP being generated, the calcium transport proteins stop pumping calcium ions into the sarcoplasmic reticulum and the calcium ions gradually leak out. This causes the myosin proteins to grab the actin and pull once, but without further supply of ATP, cannot release and pull again. The muscles therefore remain rigid in the position at death until the binding of myosin to actin begins to break down and they become loose again.[6]
Symptoms
In the event more ATP is needed from the affected pathway, the lack of it becomes an issue and symptoms develop. People with a metabolic myopathy often experience symptoms such as:
abnormal muscle fatigue (premature fatigue and/or inability to get into second wind), muscle pain (myalgia), cramping or muscle stiffness during and/or after exercise,
The degree of symptoms varies greatly from person to person and is dependent on the severity of enzymatic or transport protein defect. In extreme cases it can lead to rhabdomyolysis.[18] The symptoms experienced also depend on which metabolic pathway is impaired, as different metabolic pathways produce ATP at different time periods during activity and rest, as well as the type of activity (anaerobic or aerobic) and its intensity (level of ATP consumption).
A majority of patients with metabolic myopathies have dynamic rather than static findings, typically experiencing exercise intolerance, muscle pain, and cramps with exercise rather than fixed muscle weakness.[1][19] However, a minority of metabolic myopathies have fixed muscular weakness rather than exercise intolerance, imitating an inflammatory myopathy or limb girdle muscular dystrophy. It is uncommon that both static and dynamic signs predominate.[1][19]
Types
Metabolic myopathies are generally caused by an inherited genetic mutation, an inborn error of metabolism. (In livestock, an acquired environmental GSD is caused by intoxication with the alkaloid castanospermine.)[20] Metabolic myopathies cause the underproduction of adenosine triphosphate (ATP) within the muscle cell.[21]
The genetic mutation typically has an autosomal recessive hereditary pattern making it fairly rare to inherit, and even more rarely it can be caused by a random de novo genetic mutation, or autosomal dominant, X-linked, or mitochondrial.[1] Metabolic myopathies are categorized by the metabolic pathway to which the deficient enzyme or transport protein belongs. The main categories of metabolic myopathies are listed below:[22]
Fatty acid metabolism disorder (fatty acid oxidation disorder, FAOD)—defect in fat (lipid) metabolism, anywhere along the pathway, starting from entering the muscle cell and ending at converting fatty acids into acetyl-CoA within the mitochondrion. The deficiency occurs in the cell membrane, cytosol, mitochondrial membrane, or within the mitochondrion of the muscle cell.
The symptoms of a metabolic myopathy can be easily confused with the symptoms of another disease. As genetic sequencing research progresses, a non-invasive neuromuscular panel DNA test can help make a diagnosis. Whole genome sequencing is required in more complex cases.[1] If the DNA test is inconclusive (negative or VUS), then a muscle biopsy is necessary for an accurate diagnosis. In mitochondrial myopathies involving a single mtDNA deletion, DNA would have to be tested from affected muscle tissue rather than saliva or blood as unaffected tissues would show normal or near normal levels of mtDNA.[1][24][25]
A blood test for creatine kinase (CK) can be done under normal circumstances to test for signs of tissue breakdown, or with an added cardio portion that can indicate if muscle breakdown is occurring. In metabolic myopathies, baseline CK is either normal or elevated.[8] An electromyography (EMG) test is sometimes taken in order to rule out other disorders if the cause of fatigue is unknown.[21] In metabolic myopathies, the EMG is either normal or myopathic, but spontaneous activity is usually absent.[8]
An exercise stress test can be used to determine an inappropriate rapid heart rate (sinus tachycardia) response to exercise, which is seen in GSD-V, other glycogenoses, and mitochondrial myopathies.[7][9] A 12 Minutes Walk Test (12MWT) can also be used to determine "second wind" which is also seen in McArdle disease (GSD-V) and phosphoglucomutase deficiency (PGM1-CDG/CDG1T/GSD-XIV).[7][26]
A cardiopulmonary exercise test can measure both heart rate and breathing, to evaluate the oxygen cost (∆V’O2/∆Work-Rate) during incremental exercise. In both glycogenoses and mitochondrial myopathies, patients displayed an increased oxygen cost during exercise compared to control subjects; and therefore, can perform less work for a given V̇O2 consumption during submaximal daily life exercises.[9][27]
In fatty acid oxidation disorders (FAOD), while at rest, some exhibit cardiac arrhythmia (commonly various forms of tachycardia, but more rarely, conduction disorders or acute bradycardia); while others have a normal heart rhythm.[28]
Some GSDs and a mitochondrial myopathy are known to have a pseudoathletic appearance. McArdle disease (GSD-V) and late-onset Pompe disease (GSD-II) are known to have hypertrophy, particularly of the calf muscles.[14][15] Cori/Forbes disease (GSD-III) is known to have hypertrophy of the sternocleidomastoid, trapezius, quadriceps, and thigh muscles.[13][29][30][31] Muscular dystrophy, limb-girdle, type 1H (which as of 2017 was excluded from LGMD for showing signs on muscle biopsy as being a mitochondrial myopathy, but not yet assigned new nomenclature)[32] is also known to have hypertrophy of the calf muscles.[33]Hereditary myopathy with lactic acidosis (HML), another mitochondrial myopathy, also has hypertrophy of the calf muscles in some.[16][34]
Blood test may show a disturbance in pH, with lactic acidosis (low pH) in mitochondrial myopathies either at rest or exercise-induced.[35] Glycogen storage diseases may show transient exercise-induced alkalosis (high pH), hyperammonemia, and myogenic hyperuricemia.[36][37][38][39][40] During a non-ischemic forearm exercise test, in GSDs the plasma lactate typically fails to rise (and may fall below resting levels); except for a few GSDs such as phosphoglucomutase deficiency (GSD-XIV),[40] deficiency of functioning myophosphorylase-a (autosomal dominant PYGM),[41] phosphorylase-b kinase deficiency (GSD-IXd), and Pompe disease (GSD-II) where lactate production is normal.[2] In myoadenylate deaminase deficiency (AMPD1 deficiency), there is no rise in ammonia.[2] Some fatty acid oxidation disorders show lactic acidosis, hypoketotic hypoglycaemia and hyperammonemia, while others are asymptomatic.[2][42][43]
Differentiating between different types of metabolic myopathies can be difficult due to the similar symptoms of each type such as myoglobinuria and exercise intolerance. It has to be determined whether the patient has fixed (static) or exercise-induced (dynamic) manifestations; and if exercise-related, what kind of exercise, before extensive exercise-related lab testing is done to determine the underlying cause.[22]
which circumstances constitute anaerobic exercise (blood flow restricted by contracted muscles, insufficient oxygen and blood borne fuels, particularly isometric exercise, as well as sudden increased intensity) versus aerobic exercise (blood flow unrestricted);
anaerobic metabolism (phosphagen system and anaerobic glycolysis - ATP produced without oxygen, regardless of adequate blood flow or not, quickly produces ATP which is useful in high-intensity activity and the beginning of any activity) versus aerobic metabolism (oxidative phosphorylation - ATP produced with oxygen, adequate blood flow required, slow to produce ATP but produces for longer and high yield);
how long does each source take to start producing ATP;
how long does each source continue to produce ATP;
how long does each source take to replenish;
how much ATP can each source generate;
and which fuel source is primarily used given the intensity of the activity.
For example, leisurely-paced walking and fast-paced walking on level ground (no incline) are both aerobic, but fast-paced walking relies on more muscle glycogen because of the higher intensity (which would cause exercise intolerance symptoms in those with muscle glycogenoses that hadn't yet achieved "second wind").[11][7][17][45]
When walking at a leisurely pace on level ground (no incline), but there is loose gravel or sand, long grass, snow, mud, or walking into a headwind, that added resistance (requiring more effort) makes the activity more reliant on muscle glycogen also.[7][17] These and other surfaces, such as ice, can make you tense your muscles (which is anearobic requiring muscle glycogen) as you protect yourself from slipping or falling.[7][17]
Those with muscle glycogenoses can maintain a healthy life of exercise by learning activity adaptations, utilizing the bioenergetic systems that are available to them. Depending on the type of activity and whether they are in second wind, they slow their pace or rest briefly when need be, to make sure not to empty their "ATP reservoir."[7][17]
Rapid (tachypnea) and commonly heavy breathing (hyperpnea) with exercise (exercise hyperventilation);
Inappropriate rapid heart rate response to exercise;
Muscle fatigue, pain (myalgia), and cramps with exercise;
Sucrose taken shortly before exercise mitigates symptoms in glycogenolytic defects (e.g. GSD-V) or worsens symptoms in glycolytic defects (e.g. GSD-VII);
Ketosis improves symptoms;
May have a pseudoathletic appearance (particularly of the calf muscles);
Rhabdomyolysis and myoglobinuria possible;
Baseline creatine kinase normal or elevated;
Plasma pH disturbance of alkalosis during exercise;
Exercise-induced hyperammonemia and myogenic hyperuricemia with lactate failing to rise (rarely normal).
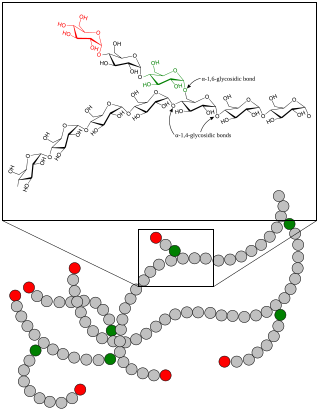
Glycogen storage disease
Treatment
Metabolic myopathies have varying levels of symptoms, being most severe when developed during infancy. Those who do not develop a form of a metabolic myopathy until they are in their young adult or adult life tend to have more treatable symptoms that can be helped with a change in diet and exercise.[18] It might be more accurate to say that metabolic myopathies described as adult-onset, it isn't necessarily that they didn't develop in infancy (they are inborn—from birth—errors of metabolism) but that they didn't display severe enough symptoms to warrant the attention of medical professionals until their adult years (severe symptoms such as rhabdomyolysis, fixed muscle weakness due to years of repetitive injury, or the de-conditioning of muscles from a more sedentary adult lifestyle which exacerbated symptoms).
Due to the rare nature of these diseases, it is very common to be misdiagnosed, even misdiagnosed multiple times.[11][48][44][49] Once a correct diagnosis has been made, in adult years, looking back symptoms were present since childhood, but either brushed-off as growing pains, laziness, or told that they just needed to exercise more.[44][48][11] It is especially difficult to get a diagnosis when symptoms are dynamic (exercise-induced), such as in muscle glycogenoses.[11][19][44] Sitting in a doctor's office (at rest) or doing movements that only last a few seconds (within the time limit of the phosphagen system) the patient wouldn't display any noticeable abnormalities (such as muscle fatigue, cramping, or breathlessness).
A brief or only mildly elevated heart rate (heart rate taken while sitting down after recently walking across the room or getting up on the examination table) might be assumed to be due to anxiety or illness rather than exercise-induced inappropriate rapid heart rate due to an ATP shortage in the muscle cells. In the absence of severe symptoms (such as hepatomegaly, cardiomyopathy, hypoglycemia, lactic acidosis, myoglobinuria, rhabdomyolysis, acute compartment syndrome or renal failure), it is understandable that a disease would not be noticed by medical professionals for years, when at rest the patient appears completely normal.
Depending on what enzyme is affected, a high-protein or low-fat diet may be recommended along with mild exercise. It is important for people with metabolic myopathies to consult with their doctors for a treatment plan in order to prevent acute muscle breakdowns while exercising that lead to the release of muscle proteins into the bloodstream that can cause kidney damage.[21]
A ketogenic diet has a remarkable effect on CNS-symptoms in PDH-deficiency and has also been tried in complex I deficiency.[50] A ketogenic diet has demonstrated beneficial for McArdle disease (GSD-V) as ketones readily convert to acetyl CoA for oxidative phosphorylation, whereas free fatty acids take a few minutes to convert into acetyl CoA.[47] As of 2022, another study on a ketogenic diet and McArdle disease (GSD-V) is underway.[51]
For McArdle disease (GSD-V), regular aerobic exercise utilizing "second wind" to enable the muscles to become aerobically conditioned, as well as anaerobic exercise that follows the activity adaptations so as not to cause muscle injury, helps to improve exercise intolerance symptoms and maintain overall health.[7][11][52][53] Studies have shown that regular low-moderate aerobic exercise increases peak power output, increases peak oxygen uptake (VO2peak), lowers heart rate, and lowers serum CK in individuals with McArdle disease.[52][53]
Regardless of whether the patient experiences symptoms of muscle pain, muscle fatigue, or cramping, the phenomenon of second wind having been achieved is demonstrable by the sign of an increased heart rate dropping while maintaining the same speed on the treadmill.[53][44] Inactive patients experienced second wind, demonstrated through relief of typical symptoms and the sign of an increased heart rate dropping, while performing low-moderate aerobic exercise (walking or brisk walking).[53][44] Conversely, patients that were regularly active did not experience the typical symptoms during low-moderate aerobic exercise (walking or brisk walking), but still demonstrated second wind by the sign of an increased heart rate dropping.[53][44] For the regularly active patients, it took more strenuous exercise (very brisk walking/jogging or bicycling) for them to experience both the typical symptoms and relief thereof, along with the sign of an increased heart rate dropping, demonstrating second wind.[53][44]
Society and culture
The American TV show, Diagnosis, in episode 1 Detective Work, an athletic 23-year-old nursing student, Angel Parker, experiences episodes of extreme muscle fatigue, pain and cramping after prolonged exercise, sometimes followed by dark urine (myoglobinuria) and elevated CK (rhabdomyolysis). These episodes can leave her immobile for hours. She had experienced muscle pains since childhood, passed-off as "growing pains." Her first hospitalization was at age 14, for intolerable leg pain that woke her in the middle of the night, sobbing uncontrollably from the intense pain. After multiple hospitalizations for myoglobinuria and rhabdomyolysis, many misdiagnoses, and many different doctors and tests that were unable to give an explanation for her symptoms, through the assistance of the show genetic sequencing confirmed that she had CPT-II deficiency (a fatty-acid metabolism disorder).[54][55]
In 2010, Walk over Wales (WoW), the first walking course for those with muscle glycogen storage disease occurred to teach participants activity adaptations, swap notes, and raise public awareness. Led by Andrew Wakelin, participants came from around the world, including Stacey Reason, Dan Chambers, Andy Williams, Charlton Thear and Dr. Ros Quinlivan.[56][57] From Great Orme to Cardiff Bay, in 32 days they walked 210 miles (338 km) and ascended approximately 35,000 feet (10,700 metres) through Snowdonia National Park, the Cambrian Mountains, and Brecon Beacons National Park. There have been many walking courses over the years since, growing in participation at international locations.[58][59]
Mattie J.T. Stepanek (1990-2004), an American poet and peace activist, died aged 13 from dysautonomic mitochondrial myopathy, an inherited disease that causes muscle weakness and impairs heart rate, breathing, blood pressure, and digestion. Predeceased by his older siblings, he was survived by his mother (then aged 44), who was diagnosed in 1992 with a late-onset form of the same disease.[60]
Charles Darwin (1809-1882), an English naturalist and biologist, suffered from a chronic illness that has been speculated to have been MELAS syndrome (a mitochondrial myopathy).[61]
Kocher–Debré–Semelaigne syndrome(KDSS) is hypothyroidism in infancy or childhood characterised by lower extremity or generalized muscular hypertrophy (Herculean appearance), myxoedema, short stature, and cognitive impairment.
Adenosine monophosphate deaminase deficiency type 1 or AMPD1, is a human metabolic disorder in which the body consistently lacks the enzyme AMP deaminase, in sufficient quantities. This may result in exercise intolerance, muscle pain and muscle cramping. The disease was formerly known as myoadenylate deaminase deficiency (MADD).
Glycogen storage disease type V, also known as McArdle's disease, is a metabolic disorder, one of the metabolic myopathies, more specifically a muscle glycogen storage disease, caused by a deficiency of myophosphorylase. Its incidence is reported as one in 100,000, roughly the same as glycogen storage disease type I.
A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.
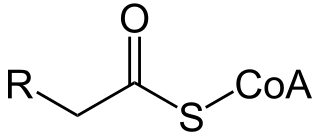
Carnitine is a quaternary ammonium compound involved in metabolism in most mammals, plants, and some bacteria. In support of energy metabolism, carnitine transports long-chain fatty acids from the cytosol into mitochondria to be oxidized for free energy production, and also participates in removing products of metabolism from cells. Given its key metabolic roles, carnitine is concentrated in tissues like skeletal and cardiac muscle that metabolize fatty acids as an energy source. Generally individuals, including strict vegetarians, synthesize enough L-carnitine in vivo.
Alkalosis is the result of a process reducing hydrogen ion concentration of arterial blood plasma (alkalemia). In contrast to acidemia, alkalemia occurs when the serum pH is higher than normal. Alkalosis is usually divided into the categories of respiratory alkalosis and metabolic alkalosis or a combined respiratory/metabolic alkalosis.
Phosphoglucomutase is an enzyme that transfers a phosphate group on an α-D-glucose monomer from the 1 to the 6 position in the forward direction or the 6 to the 1 position in the reverse direction.
Phosphofructokinase deficiency is a rare muscular metabolic disorder, with an autosomal recessive inheritance pattern.
Exercise intolerance is a condition of inability or decreased ability to perform physical exercise at the normally expected level or duration for people of that age, size, sex, and muscle mass. It also includes experiences of unusually severe post-exercise pain, fatigue, nausea, vomiting or other negative effects. Exercise intolerance is not a disease or syndrome in and of itself, but can result from various disorders.
In endurance sports such as road cycling and long-distance running, hitting the wall or the bonk is a condition of sudden fatigue and loss of energy which is caused by the depletion of glycogen stores in the liver and muscles. Milder instances can be remedied by brief rest and the ingestion of food or drinks containing carbohydrates. Otherwise, it can remedied by attaining second wind by either resting for approximately 10 minutes or by slowing down considerably and increasing speed slowly over a period of 10 minutes. Ten minutes is approximately the time that it takes for free fatty acids to sufficiently produce ATP in response to increased demand.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA. Acetyl-CoA enters the citric acid cycle, generating NADH and FADH2, which are electron carriers used in the electron transport chain. It is named as such because the beta carbon of the fatty acid chain undergoes oxidation and is converted to a carbonyl group to start the cycle all over again. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.
Sinus tachycardia is a sinus rhythm of the heart, with an increased rate of electrical discharge from the sinoatrial node, resulting in a tachycardia, a heart rate that is higher than the upper limit of normal.
Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.
Myophosphorylase or glycogen phosphorylase, muscle associated (PYGM) is the muscle isoform of the enzyme glycogen phosphorylase and is encoded by the PYGM gene. This enzyme helps break down glycogen into glucose-1-phosphate, so it can be used within the muscle cell. Mutations in this gene are associated with McArdle disease, a glycogen storage disease of muscle.
Bioenergetic systems are metabolic processes that relate to the flow of energy in living organisms. Those processes convert energy into adenosine triphosphate (ATP), which is the form suitable for muscular activity. There are two main forms of synthesis of ATP: aerobic, which uses oxygen from the bloodstream, and anaerobic, which does not. Bioenergetics is the field of biology that studies bioenergetic systems.
Second wind is a phenomenon in endurance sports, such as marathons or road running, whereby an athlete who is out of breath and too tired to continue, finds the strength to press on at top performance with less exertion. The feeling may be similar to that of a "runner's high", the most obvious difference being that the runner's high occurs after the race is over. In muscle glycogenoses, an inborn error of carbohydrate metabolism impairs either the formation or utilization of muscle glycogen. As such, those with muscle glycogenoses do not need to do prolonged exercise to experience "hitting the wall". Instead, signs of exercise intolerance, such as an inappropriate rapid heart rate response to exercise, are experienced from the beginning of an activity, and some muscle GSDs can achieve second wind within about 10 minutes from the beginning of the aerobic activity, such as walking. (See below in pathology).
Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.
Hoffmann syndrome is a rare form of hypothyroid myopathy and is not to be confused with Werdnig-Hoffmann disease.
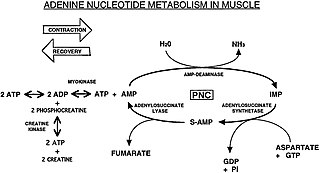
The Purine Nucleotide Cycle is a metabolic pathway in protein metabolism requiring the amino acids aspartate and glutamate. The cycle is used to regulate the levels of adenine nucleotides, in which ammonia and fumarate are generated. AMP converts into IMP and the byproduct ammonia. IMP converts to S-AMP (adenylosuccinate), which then converts to AMP and the byproduct fumarate. The fumarate goes on to produce ATP (energy) via oxidative phosphorylation as it enters the Krebs cycle and then the electron transport chain. Lowenstein first described this pathway and outlined its importance in processes including amino acid catabolism and regulation of flux through glycolysis and the Krebs cycle.
1 2 Marbini A, Gemignani F, Saccardi F, Rimoldi M (October 1989). "Debrancher deficiency neuromuscular disorder with pseudohypertrophy in two brothers". Journal of Neurology. 236 (7): 418–420. doi:10.1007/BF00314902. PMID2809644. S2CID21158814.
1 2 Løkken N, Hansen KK, Storgaard JH, Ørngreen MC, Quinlivan R, Vissing J. Titrating a modified ketogenic diet for patients with McArdle disease: A pilot study. J Inherit Metab Dis. 2020 Jul;43(4):778-786. doi:10.1002/jimd.12223 Epub 2020 Feb 24. PMID 32060930.
↑ Das AM, Steuerwald U, Illsinger S. Inborn errors of energy metabolism associated with myopathies. J Biomed Biotechnol. 2010;2010:340849. doi:10.1155/2010/340849 Epub 2010 May 26. PMID 20589068; PMCID: PMC2877206.
↑ Clinical trial number NCT04694547 for "Ketogenic Diet Survey in Patients With McArdle Disease (GSDV)" at ClinicalTrials.gov
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.