Deoxyribonucleic acid is a polymer composed of two polynucleotide chains that coil around each other to form a double helix. The polymer carries genetic instructions for the development, functioning, growth and reproduction of all known organisms and many viruses. DNA and ribonucleic acid (RNA) are nucleic acids. Alongside proteins, lipids and complex carbohydrates (polysaccharides), nucleic acids are one of the four major types of macromolecules that are essential for all known forms of life.

Genetics is the study of genes, genetic variation, and heredity in organisms. It is an important branch in biology because heredity is vital to organisms' evolution. Gregor Mendel, a Moravian Augustinian friar working in the 19th century in Brno, was the first to study genetics scientifically. Mendel studied "trait inheritance", patterns in the way traits are handed down from parents to offspring over time. He observed that organisms inherit traits by way of discrete "units of inheritance". This term, still used today, is a somewhat ambiguous definition of what is referred to as a gene.

A mitochondrion is an organelle found in the cells of most eukaryotes, such as animals, plants and fungi. Mitochondria have a double membrane structure and use aerobic respiration to generate adenosine triphosphate (ATP), which is used throughout the cell as a source of chemical energy. They were discovered by Albert von Kölliker in 1857 in the voluntary muscles of insects. The term mitochondrion was coined by Carl Benda in 1898. The mitochondrion is popularly nicknamed the "powerhouse of the cell", a phrase coined by Philip Siekevitz in a 1957 article of the same name.
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.

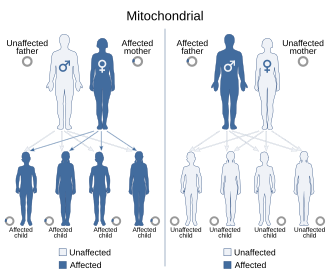
Mitochondrial DNA is the DNA located in mitochondria, cellular organelles within eukaryotic cells that convert chemical energy from food into a form that cells can use, such as adenosine triphosphate (ATP). Mitochondrial DNA is only a small portion of the DNA in a eukaryotic cell; most of the DNA can be found in the cell nucleus and, in plants and algae, also in plastids such as chloroplasts.
Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.
Nuclear DNA (nDNA), or nuclear deoxyribonucleic acid, is the DNA contained within each cell nucleus of a eukaryotic organism. It encodes for the majority of the genome in eukaryotes, with mitochondrial DNA and plastid DNA coding for the rest. It adheres to Mendelian inheritance, with information coming from two parents, one male and one female—rather than matrilineally as in mitochondrial DNA.
Genetics, a discipline of biology, is the science of heredity and variation in living organisms.

A nuclear gene is a gene that has its DNA nucleotide sequence physically situated within the cell nucleus of a eukaryotic organism. This term is employed to differentiate nuclear genes, which are located in the cell nucleus, from genes that are found in mitochondria or chloroplasts. The vast majority of genes in eukaryotes are nuclear.
Extrachromosomal DNA is any DNA that is found off the chromosomes, either inside or outside the nucleus of a cell. Most DNA in an individual genome is found in chromosomes contained in the nucleus. Multiple forms of extrachromosomal DNA exist, and, while some of these serve important biological functions, they can also play a role in diseases such as cancer.
In biology, the word gene has two meanings. The Mendelian gene is a basic unit of heredity. The molecular gene is a sequence of nucleotides in DNA, that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
Nuclear mitochondrial DNA (NUMT) segments or genetic loci describe a transposition of any type of cytoplasmic mitochondrial DNA into the nuclear genome of eukaryotic organisms.

Cytochrome c oxidase I (COX1) also known as mitochondrially encoded cytochrome c oxidase I (MT-CO1) is a protein that is encoded by the MT-CO1 gene in eukaryotes. The gene is also called COX1, CO1, or COI. Cytochrome c oxidase I is the main subunit of the cytochrome c oxidase complex. In humans, mutations in MT-CO1 have been associated with Leber's hereditary optic neuropathy (LHON), acquired idiopathic sideroblastic anemia, Complex IV deficiency, colorectal cancer, sensorineural deafness, and recurrent myoglobinuria.
In molecular biology, a displacement loop or D-loop is a DNA structure where the two strands of a double-stranded DNA molecule are separated for a stretch and held apart by a third strand of DNA. An R-loop is similar to a D-loop, but in that case the third strand is RNA rather than DNA. The third strand has a base sequence which is complementary to one of the main strands and pairs with it, thus displacing the other complementary main strand in the region. Within that region the structure is thus a form of triple-stranded DNA. A diagram in the paper introducing the term illustrated the D-loop with a shape resembling a capital "D", where the displaced strand formed the loop of the "D".
Genome instability refers to a high frequency of mutations within the genome of a cellular lineage. These mutations can include changes in nucleic acid sequences, chromosomal rearrangements or aneuploidy. Genome instability does occur in bacteria. In multicellular organisms genome instability is central to carcinogenesis, and in humans it is also a factor in some neurodegenerative diseases such as amyotrophic lateral sclerosis or the neuromuscular disease myotonic dystrophy.

The mitochondrial ribosome, or mitoribosome, is a protein complex that is active in mitochondria and functions as a riboprotein for translating mitochondrial mRNAs encoded in mtDNA. The mitoribosome is attached to the inner mitochondrial membrane. Mitoribosomes, like cytoplasmic ribosomes, consist of two subunits — large (mt-LSU) and small (mt-SSU). Mitoribosomes consist of several specific proteins and fewer rRNAs. While mitochondrial rRNAs are encoded in the mitochondrial genome, the proteins that make up mitoribosomes are encoded in the nucleus and assembled by cytoplasmic ribosomes before being implanted into the mitochondria.
A linear chromosome is a chromosome which is linear in shape, and contains terminal ends. In most eukaryotic cells, DNA is arranged in multiple linear chromosomes. In contrast, most prokaryotic cells generally contain a singular circular chromosome.
This glossary of cellular and molecular biology is a list of definitions of terms and concepts commonly used in the study of cell biology, molecular biology, and related disciplines, including molecular genetics, biochemistry, and microbiology. It is split across two articles:
This glossary of cellular and molecular biology is a list of definitions of terms and concepts commonly used in the study of cell biology, molecular biology, and related disciplines, including genetics, biochemistry, and microbiology. It is split across two articles: