
Hemolysis or haemolysis, also known by several other names, is the rupturing (lysis) of red blood cells (erythrocytes) and the release of their contents (cytoplasm) into surrounding fluid. Hemolysis may occur in vivo or in vitro.

Immune thrombocytopenic purpura (ITP), also known as idiopathic thrombocytopenic purpura or immune thrombocytopenia, is a type of thrombocytopenic purpura characterized by a low platelet count in the absence of other causes, and accompanied by a red-purple rash called purpura. It leads to an increased risk of bleeding. ITP manifests in two distinct clinical syndromes: an acute form observed in children, and chronic conditions observed in adults. The acute form often follows an infection and typically resolves within two months, while chronic immune thrombocytopenia persists for longer than six months and its specific cause is unknown.
Microangiopathic hemolytic anemia (MAHA) is a microangiopathic subgroup of hemolytic anemia caused by factors in the small blood vessels. It is identified by the finding of anemia and schistocytes on microscopy of the blood film.
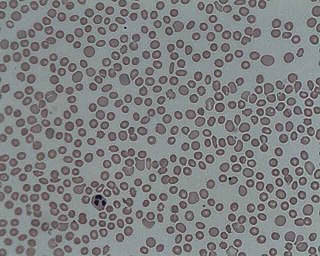
In hematology, thrombocytopenia is a condition characterized by abnormally low levels of platelets in the blood. Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.

Hemolytic–uremic syndrome (HUS) is a group of blood disorders characterized by low red blood cells, acute kidney injury, and low platelets. Initial symptoms typically include bloody diarrhea, fever, vomiting, and weakness. Kidney problems and low platelets then occur as the diarrhea progresses. Children are more commonly affected, but most children recover without permanent damage to their health, although some children may have serious and sometimes life-threatening complications. Adults, especially the elderly, may present a more complicated presentation. Complications may include neurological problems and heart failure.

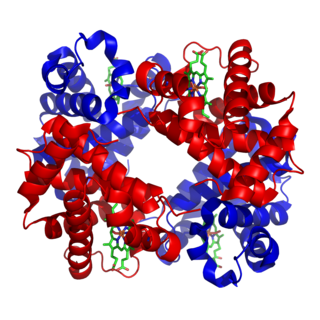
Von Willebrand factor (VWF) is a blood glycoprotein that promotes hemostasis, specifically, platelet adhesion. It is deficient and/or defective in von Willebrand disease and is involved in many other diseases, including thrombotic thrombocytopenic purpura, Heyde's syndrome, and possibly hemolytic–uremic syndrome. Increased plasma levels in many cardiovascular, neoplastic, metabolic, and connective tissue diseases are presumed to arise from adverse changes to the endothelium, and may predict an increased risk of thrombosis.
Hemoglobinuria is a condition in which the oxygen transport protein hemoglobin is found in abnormally high concentrations in the urine. The condition is caused by excessive intravascular hemolysis, in which large numbers of red blood cells (RBCs) are destroyed, thereby releasing free hemoglobin into the plasma. Excess hemoglobin is filtered by the kidneys, which excrete it into the urine, giving urine a purple color. Hemoglobinuria can lead to acute tubular necrosis which is an uncommon cause of a death of uni-traumatic patients recovering in the ICU.
HELLP syndrome is a complication of pregnancy; the acronym stands for hemolysis, elevated liver enzymes, and low platelet count. It usually begins during the last three months of pregnancy or shortly after childbirth. Symptoms may include feeling tired, retaining fluid, headache, nausea, upper right abdominal pain, blurry vision, nosebleeds, and seizures. Complications may include disseminated intravascular coagulation, placental abruption, and kidney failure.
Evans syndrome is an autoimmune disease in which an individual's immune system attacks their own red blood cells and platelets, the syndrome can include immune neutropenia. These immune cytopenias may occur simultaneously or sequentially.

A schistocyte or schizocyte is a fragmented part of a red blood cell. Schistocytes are typically irregularly shaped, jagged, and have two pointed ends.

ADAMTS13 —also known as von Willebrand factor-cleaving protease (VWFCP)—is a zinc-containing metalloprotease enzyme that cleaves von Willebrand factor (vWf), a large protein involved in blood clotting. It is secreted into the blood and degrades large vWf multimers, decreasing their activity, hence ADAMTS13 acts to reduces thrombus formation.
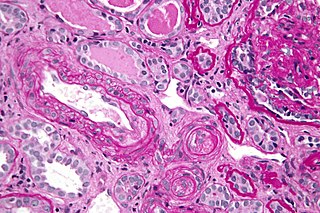
Thrombotic microangiopathy (TMA) is a pathology that results in thrombosis in capillaries and arterioles, due to an endothelial injury. It may be seen in association with thrombocytopenia, anemia, purpura and kidney failure.
Hematologic diseases are disorders which primarily affect the blood & blood-forming organs. Hematologic diseases include rare genetic disorders, anemia, HIV, sickle cell disease & complications from chemotherapy or transfusions.
The term cryosupernatant refers to plasma from which the cryoprecipitate has been removed. It is used to treat thrombocytopenic purpura.
Caplacizumab is a bivalent single-domain antibody (VHH) designed for the treatment of thrombotic thrombocytopenic purpura (TTP) and thrombosis.
Atypical hemolytic uremic syndrome (aHUS), also known as complement-mediated hemolytic uremic syndrome, is an extremely rare, life-threatening, progressive disease that frequently has a genetic component. In most cases it can be effectively controlled by interruption of the complement cascade. Particular monoclonal antibodies, discussed later in the article, have proven efficacy in many cases.
Upshaw–Schulman syndrome (USS) is the recessively inherited form of thrombotic thrombocytopenic purpura (TTP), a rare and complex blood coagulation disease. USS is caused by the absence of the ADAMTS13 protease resulting in the persistence of ultra large von Willebrand factor multimers (ULVWF), causing episodes of acute thrombotic microangiopathy with disseminated multiple small vessel obstructions. These obstructions deprive downstream tissues from blood and oxygen, which can result in tissue damage and death. The presentation of an acute USS episode is variable but usually associated with thrombocytopenia, microangiopathic hemolytic anemia (MAHA) with schistocytes on the peripheral blood smear, fever and signs of ischemic organ damage in the brain, kidney and heart.
Eli Moschcowitz was an American doctor best known for his role in discovering thrombotic thrombocytopenic purpura (TTP), which was originally called "Moschcowitz syndrome". He is also known for having an early role in the development of psychosomatic medicine.
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening disorder characterized by thrombocytopenia and microangiopathic hemolytic anemia accompanied by variable neurological dysfunction, kidney failure, and fever. It is caused by severely reduced activity of the von Willebrand factor-cleaving protease ADAMTS13. Hereditary TTP, caused by ADAMTS13 gene mutations, is much less common.
Hemolytic jaundice, also known as prehepatic jaundice, is a type of jaundice arising from hemolysis or excessive destruction of red blood cells, when the byproduct bilirubin is not excreted by the hepatic cells quickly enough. Unless the patient is concurrently affected by hepatic dysfunctions or is experiencing hepatocellular damage, the liver does not contribute to this type of jaundice.

